 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Memórias do Instituto Oswaldo Cruz, Vol. 105, No. 2, 2010, pp. 233-238 TECHNICAL NOTE In vitro and in vivo experimental models for drug screening and development for Chagas disease Alvaro José RomanhaI, II, +; Solange Lisboa de CastroI, III; Maria de Nazaré Correia SoeiroI, III; Joseli Lannes-VieiraI, III; Isabela RibeiroIV; André TalvaniV; Bernadette BourdinIV; Bethania BlumIV; Bianca OlivieriI, III; Carlos ZaniI, II; Carmenza SpadaforaVI; Egler ChiariVII; Eric ChatelainIV; Gabriela ChavesVIII; José Eduardo CalzadaIX; Juan Manuel BustamanteX; Lucio H Freitas-JuniorXI; Luz I RomeroVI, IX; Maria Terezinha BahiaV; Michel LotrowskaIV; Milena SoaresXII; Sonia Gumes AndradeI, XII, Tanya ArmstrongXIII; Wim DegraveI, III; Zilton de Araújo AndradeI, XII IPrograma Integrado de Doença de Chagas-Fiocruz, Rio de Janeiro, RJ, Brasil Received 22 October 2009 Financial support: Fiocruz, DNDi Code Number: oc10041 ABSTRACT Chagas disease, a neglected illness, affects nearly 12-14 million people in endemic areas of Latin America. Although the occurrence of acute cases sharply has declined due to Southern Cone Initiative efforts to control vector transmission, there still remain serious challenges, including the maintenance of sustainable public policies for Chagas disease control and the urgent need for better drugs to treat chagasic patients. Since the introduction of benznidazole and nifurtimox approximately 40 years ago, many natural and synthetic compounds have been assayed against Trypanosoma cruzi, yet only a few compounds have advanced to clinical trials. This reflects, at least in part, the lack of consensus regarding appropriate in vitro and in vivo screening protocols as well as the lack of biomarkers for treating parasitaemia. The development of more effective drugs requires (i) the identification and validation of parasite targets, (ii) compounds to be screened against the targets or the whole parasite and (iii) a panel of minimum standardised procedures to advance leading compounds to clinical trials. This third aim was the topic of the workshop entitled Experimental Models in Drug Screening and Development for Chagas Disease, held in Rio de Janeiro, Brazil, on the 25th and 26th of November 2008 by the Fiocruz Program for Research and Technological Development on Chagas Disease and Drugs for Neglected Diseases Initiative. During the meeting, the minimum steps, requirements and decision gates for the determination of the efficacy of novel drugs for T. cruzi control were evaluated by interdisciplinary experts and an in vitro and in vivo flowchart was designed to serve as a general and standardised protocol for screening potential drugs for the treatment of Chagas disease. Key words: Trypanosoma cruzi - Chagas disease treatment - drug screening - benznidazole In 1975, Zigman Brener said: "A one-shot inexpensive, nontoxic drug to be used in individual cases as well as for preventing Chagas disease transmission is still a vague dream. There is, then, plenty of room for new active compounds against Trypanosoma cruzi." There is a continuous demand for novel therapies for tropical diseases, including malaria and Chagas disease. The lack of consensus regarding in vitro and in vivo screening protocols and the absence of disease markers have hampered the discovery of new drugs for treating Chagas disease. Accordingly, the identification of a screening panel that allows for the assessment of a compound as a potential therapeutic and advances the compound from the bench to clinical trials in as few steps as necessary is critical. This type of panel was the topic of discussion at the workshop entitled Experimental Models in Drug Screening and Development for Chagas Disease, which was held in Rio de Janeiro, Brazil, on the 25th and 26th of November 2008 and presented by the Fiocruz Program for Research and Technological Development on Chagas Disease (PIDC/Fiocruz) and the Drugs for Neglected Diseases Initiative (DNDi). PIDC was launched in 2000 and consists of five networks involving 34 laboratories at Fiocruz. The aim of the PIDC is to promote the development of research and technological projects focused on Chagas disease and to enhance the cost-effectiveness of such projects. DNDi is a non-profit product development partnership founded in 2003 by seven organisations (Fiocruz, the Indian Council for Medical Research, the Kenya Medical Research Institute, the Ministry of Health of Malaysia, France's Pasteur Institute, Médecins sans Frontières and the WHO/TDR). The mission of the DNDi is to develop new treatments for neglected kinetoplastid diseases, including Chagas disease. Chagas disease is caused by the intracellular parasite Trypanosoma cruzi. This disease has two sequential clinical phases: the acute phase, which is usually asymptomatic and starts soon after parasite infection, lasting up to a few weeks and the chronic phase, in which 30-40% of infected patients develop cardiac and/or digestive damage after a silent period lasting from several years to decades (Chagas 1916, Laranja et al. 1956). Although vectorial and transfusional transmission have declined sharply in the past 20 years, due to Southern Cone countries' policies (Incosul, http://www.paho.org/Spanish/ad/dpc/cd/xi-incosur.htm), several challenges still need to be overcome, including those related to public policies for sustainable disease control in the endemic areas (Dias et al. 2008). Nifurtimox (3-methyl-4-(5'-nitrofurfurylideneamine)tetrahydro-4H-1,4-tiazine-1,1-dioxide, Bayer 2502) and (Bz) (N-benzyl-2-nitroimidazole acetamide, RO7-1051), which were developed more than four decades ago (Coura & De Castro 2002), remain the current treatments for Chagas disease. These nitro-containing compounds are considered far from ideal because they cause multiple side effects and present limited efficacy, especially in patients with the chronic form of the disease (Rocha et al. 2007, Soeiro et al. 2009). These limitations highlight the urgency for the identification and development of new trypanocidal compounds to replace these current chemotherapies. Although many in vitro and in vivo studies have been carried out in the last three decades and promising targets have been identified, only three compounds (allopurinol, itraconazole and fluconazole) have advanced to clinical trials (Soeiro & De Castro 2009). This problem may be due, at least in part, to the lack of well-established procedures and screening protocols to evaluate the trypanocidal activity of a given compound and to the lack of parasitological markers for preclinical assays. Therefore, the development and screening of trypanocidal leads will require the standardisation of assays to a general format that will be used to evaluate the efficacy of novel compounds (natural or synthetic) in an optimised, safe and reproducible manner. In this vein, relevant aspects of the parasite's biology and its pathogenicity to potential preclinical experimental hosts shall be considered. Therefore, establishing a minimum set of criteria and decision gates to guarantee reliable results that might lead to the identification of novel compounds to be tested in preclinical assays is a crucial step in developing a drug screening process. This paper provides information focused on in vitro and in vivo approaches to screening the trypanocidal activity and toxicity of natural or synthetic compounds in mammalian cells, with the aim of developing new drugs to treat Chagas disease. There are additional pharmacological, pharmacokinetic and chemical modification techniques (e.g., derivatisation, oral availability as part of the Lead Optimisation process) and safety tests that are not incorporated in this technical note but are nonetheless essential and should be evaluated in parallel or subsequent studies in order to advance a promising compound to clinical trials. The adopted strategy consisted of encouraging discussion and information exchange among worldwide experts representing academic, public, industrial and non-governmental institutions from different parts of Brazil, Australia, Korea, Panama and the United States. All the rounds of discussion were conducted based on guidelines previously identified and designed by the PIDC/Fiocruz and DNDi members. The aim of these discussions was to define the minimal steps of the process, represented as a flow chart (Figure), to be followed in demonstrating the anti-T. cruzi efficacy of compounds in preclinical trials. To help the participants reach conclusions, the discussions covered in vitro and in vivo procedures separately, as described below: In vitro procedures - (i) whole cell-based screening, including the definition of mammalian cell culture lineages, parasite strains and forms, (ii) automated procedures and (iii) cut-off definitions, such as the determination of the IC50 and selectivity index (SI) and comparison with those of benznidazole (Bz). In vivo procedures - (i) determination of acute toxicity in murine models [maximum tolerated dose (MTD)], (ii) reduction of parasitaemia in the acute phase and cut-off criteria for selection of compounds, (iii) parasitological markers in the acute phase (moderate and resistant T. cruzi stocks) and cut-off criteria for selection of compounds, (iv) are additional steps needed? Are chronic phase models and other animal models required? All proposed steps were discussed separately, taking into account the well-accepted current requirements for the development of novel anti-T. cruzi compounds (Urbina 1999, Nwaka & Hudson 2006), which include: (i) high activity against T. cruzi forms present in the mammalian hosts and against different stocks of the parasite, (ii) efficacy in both the acute and chronic phases of infection, (iii) oral administration and short treatment courses, (iv) safe for use in children and pregnant women, (v) low cost and high stability for a long shelf-life in tropical conditions and (vi) tissue accumulation and long-lasting half-life. Another crucial point, which is considered to be a basic requirement for all the biological assays and T. cruzi strain selection methods to be used in large screening programs, was the adoption of biosafety procedures (i.e., good laboratory practices and laboratory facilities and the use of collective and individual protection tools) and the capacity of the involved personnel to follow special recommendations (as established by the Centers for Disease Control and Prevention, http://www.cdc.gov/od/ohs/biosfty/bmbl4/bmbl4s3.htm) for the manipulation of drug-susceptible and drug-resistant T. cruzi strains. For in vitro models for Chagas disease drug discovery and development, what is known and what is needed? This discussion focused on parasite forms and stocks, mammalian cell lineages, toxicity assays, automation procedures and the definition of cut-offs. The discussion on the criteria to be adopted for in vitro models led to the definition of guidelines for the use of trypomastigotes and intracellular amastigotes, which are the parasite forms that are relevant to human infection. In vitro I - The panel recommended the direct analysis of compounds on T. cruzi-infected cell lineages at a single concentration, which allows for monitoring the effects on both amastigotes and trypomastigotes in just one system. The concentrations chosen for synthetic compounds and extracts of natural products were one and 10 μg/mL, respectively; these values were chosen based on the activity of the reference drug, Bz, approximating its IC50 value to be 3.8 μM. To allow for large-scale screening, the use of an automated assay using cell lineages such as Vero, HeLa, L929 and L6 was recommended, along with experimental conditions that lead to parasitisation in approximately 50% of the cells. Cultured trypomastigotes from the β-galactosidase-transfected Tulahuen strain were the recommended parasite source (Buckner et al. 1996, Bettiol et al. 2009). The infected cells will be treated for four days and the enzymatic activity will be measured at 570 nm after the addition of chlorophenol red (Appendix A). Compounds with similar or greater trypanocidal effects as compared to Bz will move on to the next phase of screening. In vitro II - The selected compounds should be evaluated by a dose-response assay to determine their IC50 values (the drug concentration that eliminates 50% of the parasites), using the same experimental model as described above and cell lines infected with cultured trypomastigotes from the β-galactosidase-transfected Tulahuen strain. The SI is determined based on the ratio of the IC50 value in the host cell divided by the IC50 value of the parasite. Only those compounds that attain an SI value greater than 50 (Nwaka & Hudson 2006) will be considered for further screening. Next, a single drug concentration equivalent to 50X the parasite IC50 of each compound will be assayed using the AlamarBlueTM protocol to evaluate the toxicity to mammalian cells (Appendix B). Compounds that lead to < 50% cell death will advance to the next stage of screening. Due to the broad spectrum of bioactivity of various drugs in different stocks of T. cruzi (Brener & Chiari 1967, Brener et al. 1976, Andrade et al. 1977, Filardi & Brener 1984, Camandaroba et al. 2003), it is important to analyse the activity of new compounds against parasites of different lineages, including strains resistant to Bz and nifurtimox (Andrade et al. 1985, Filardi & Brener 1987). However, one of the main limitations of such an analysis is the difficulty of using different subpopulations of T. cruzi, including the Y and Colombian strains transfected with β-galactosidase, other enzymes or green fluorescent protein. It was decided that at this time, the T. cruzi Tulahuen strain transfected with β-galactosidase will be used for the In vitro I and IIprotocols. However, when other automated or alternative screening systems become available, such as the one reported by McKerrow et al. (2009), it is highly recommended that this protocol be changed to include parasite stocks from different geographical regions and stocks with known resistance to Bz and nifurtimox that belong to the six discrete typing units - T. cruzi I-VI (Zingales et al. 2009). The need to establish criteria and filters that determine the decision to advance compounds from in vitro to in vivo assays is based on: (i) a reduction in the number of animals and other ethical aspects related to the use of experimental animals, (ii) infrastructure limitations, (iii) difficulty for most laboratories to perform a high number of in vivo tests, with respect to the costs and time involved and (iv) the risk of human infection. It is important to note that, although high-stringency filters were considered in the proposed screening process, data in the literature do not support a direct correlation between the in vitro and in vivo activities of compounds that have been previously assayed for the treatment of Chagas disease. For the in vivo models for Chagas disease drug discovery and development, focusing on models of acute infection, what is known and what is needed? The discussion focused on acute drug toxicity, parasite stocks, mouse lineages, evaluation criteria for parasitaemia reduction, parasitological cure markers and cut-off values. The determination of acute toxicity in vivo was recommended as the first step to exclude compounds that are toxic to the vertebrate host. Thus, the method for determining the MTD was selected because it uses one female and one male mouse per compound and thereby fulfils the requirement of reducing, refining and replacing the use of animals in toxicity testing (also known as the 3R principle), as suggested in the guidelines of the Organisation for Economic Co-operation and Development (Walum 1998). After the MTD is determined, three sequential assays are proposed: In vivo I, to evaluate the efficacy of the compound to reduce parasitaemia, In vivo II, to evaluate the efficacy of the compound to cure mice infected with the Y strain, which is moderately resistant to Bz (Filardi & Brener 1987) and In vivo III, to evaluate the efficacy of the compound to cure mice infected with the naturally Bz-resistant Colombian strain. For a compound to progress on the flow chart (Figure), it must display trypanocidal activity equal or superior to that of Bz. These models were recommended due to the fact that they use a mouse lineage with an outbred genetic background for the evaluation of compound activity. These animals display an acute infection profile with high parasitaemia and mortality levels, allowing for a comparison between the effect of the compound/extract tested and that of Bz. As a control, the infected and non-treated group should receive the same vehicle that was used for the dilution of the compound. The In vivo I protocol allows the analyses of the effect of drugs on the parasite load using Swiss female mice (18-20 g, 6 animals/group) infected with 104 bloodstream trypomastigotes from the Y strain. Animals are treated with three doses, with the highest dose set at the MTD value. The treatment is administered orally or intraperitoneally (ip) for five consecutive days, with treatment beginning five days post-infection (dpi). Only those animals that are positive for parasitaemia will be used. The following parameters are followed: (i) parasitaemia is evaluated microscopically by the Pizzi-Brener method (Brener 1962) at five, eight and 10 dpi and (ii) survival is defined as an animal living 30 days after the end of the treatment. Results from each compound tested according to these parameters will be compared to the results achieved following the same protocol dosing with 100 mg/kg/day Bz. The Y strain was chosen because it presents moderate resistance to Bz and nifurtimox (Filardi & Brener 1987) and because it has been routinely used in in vitro and in vivo studies of drug activity, which may ultimately be useful for comparing the efficacy of various compounds. Furthermore, using this strain in these assays allows for the rapid identification (less than 40 days) of the ideal dose necessary for subsequent steps in the screening process. The In vivo II protocol allows the analyses of parasitological cure during the acute phase of infection caused by the Y strain. It employs Swiss female mice, weighing 18-20 g and uses 10 animals/group, where each animal is infected with 104 bloodstream trypomastigotes and treated with the dose established in the In vivo I step. Only those animals positive for parasitaemia are used and treatment is administered orally or intraperitoneally for 20 consecutive days, beginning 5 dpi. The parameters chosen to determine the percentage of parasitological cure induced by a given compound are (i) the presence of parasitaemia at five, eight and 10 dpi, (ii) the survival of mice 30 days after initial infection and (iii) polymerase chain reaction (PCR) (Britto et al. 1993) after immunosuppression at 30 days, as previously established (Santos et al. 2008). If the performance of a given compound or extract is similar or better than that of Bz at 100 mg/kg/day following the same treatment protocol, it will advance to the next stage of the screening process. PCR was identified as one of the necessary criteria because it is more sensitive and time efficient than haemoculture (Camandaroba et al. 2003, Caldas et al. 2008a, Miyamoto et al. 2008) and it reduces the technicians' levels of exposure to the parasite. The use of cyclophosphamide to enhance the sensitivity of the parasitological cure analysis was also discussed because immunosuppressed animals that experience therapeutic failure quickly recover their high levels of parasitaemia (Bustamante et al. 2008, Santos et al. 2008). The In vivo III protocol analyses the ability of a compound to cure parasitaemia during the acute phase of infection caused by the Bz-resistant Colombian strain. This protocol employs Swiss female mice (18-20 g, 10 animals/group) infected with 5 x 103 bloodstream trypomastigotes (inoculum leading to mortality levels > 75%) and treated with the dose established in the In vivo Iprotocol. Only animals positive for parasitaemia are used in testing. Treatment is administered orally or intraperitoneally for 20 consecutive days, beginning 7-8 days dpi, which is a time point that corresponds to the onset of parasitaemia. Parasitaemia will also be assessed at 20, 25 and 30 dpi and the viability of animals will be recorded 30 days after the end of treatment. Animals that do not show a resurgence of parasitaemia post-treatment will be subjected to immunosuppression with cyclophosphamide (Genuxal), administered in three cycles of doses of 50 mg/kg of body weight for four consecutive days, with intervals of three days between each cycle. Parasitaemia will also be evaluated during this procedure and for five days following the end of the treatment and will be determined based on PCR results negative for parasitaemia (Caldas et al. 2008b). A compound will be approved if it shows similar or better results as compared to the same assay using Bz (100 mg/kg/day) following the same procedures. Due to the known difficulty of identifying compounds that will be active against different T. cruzi populations, it is important to consider the partial data collected from the In vivo II and In vivo III steps to prevent the elimination of compounds that could otherwise be excluded due to therapeutic failure and/or adverse reactions induced by the treatment. A strength of the In vivo II protocol is the fact that most researchers can work with the Y strain, whereas only certified research teams and laboratories are able to assay strains resistant to Bz and nifurtimox. Another important aspect of the in vivo screening protocols deals with the health of the experimental animals (SPF conditions), including their adequate maintenance and handling, hygiene conditions (chow changes and cage cleaning), weight standardisation and parasite inoculums. Although reports in the literature identify benefits associated with the screening of compounds in chronically infected animals, the main objective in the context of a bioassay platform is to identify trypanocidal drugs. This goal can be readily achieved using acute phase models. Therefore, the use of in vivo chronic models, which are essential and relevant for academic knowledge of different aspects of Chagas disease pathology, does not fundamentally contribute to the identification of new trypanocidal drugs. Finally, the requirements for a new drug were addressed during the meeting. They included elimination of the infection (parasitological cure) or massive reduction in parasite load, which would lead to a beneficial arrest of the pathology. Elimination is preferred because the existence of circulating parasites could promote the evolution of tissue lesions, which is characteristic of the chronic phase of the disease. Necropsies of chronic chagasic patients show that the persistence of parasitism is related to the presence of myocarditis (Chagas 1916, Reis et al. 1997) and can possibly lead to death (Z Andrade, unpublished observations). However, considering the limitations of the tools currently available to monitor parasitological cure, a massive reduction in parasite load would be considered to be an important advance towards the identification of a new trypanocidal drug. APENDIXES Appendix A: in vitro test of trypanocidal compounds adopting β-galactosidase-transfected Tulahuen T. cruzi strain - This assay is performed as previously described by Buckner et al. (1996), using T. cruzi (Tulahuen strain) expressing the Escherichia coli β-galactosidase gene. Infective trypomastigote forms are obtained through culture in monolayers of mouse L929 fibroblasts in RPMI-1640 medium (pH 7.2-7.4), without phenol red (Gibco BRL), containing 10% foetal bovine serum and 2 mM glutamine. For the bioassay, 4,000 L929 cells in 80 μL of supplemented medium are added to each well of a 96-well microtitre plate. After an overnight incubation, 40,000 trypomastigotes in 20 μL are added to the cells and the cells are incubated for 2 h. Medium containing parasites that did not penetrate the cells is replaced with 200 μL of fresh medium and the plate is incubated for an additional 48 h to establish infection. The medium is then replaced with solutions of compounds at 1.0 μg/mL or extracts of natural products at 10.0 μg/mL in fresh medium (200 μL) and the plate is incubated for 96 h at 37°C. After this period, 50 μL of 500 μM chlorophenol red glycoside in 0.5% Nonidet P40 is added to each well and the plate is incubated for 18 h at 37°C, after which the absorbance at 570 nm is measured. Controls with uninfected cells and infected cells treated with Bz or not are run in parallel. The results are expressed as the percentage of T. cruzi growth inhibition in compound-tested cells as compared to the infected cells and untreated cells. Triplicate assays are run in the same plate. Appendix B: in vitro cytotoxic test of trypanocidal compounds using AlamarBlueTM assay - For this bioassay, 4,000 mammalian cells in 200 μL of RPMI-1640 medium (pH 7.2-7.4) (Gibco BRL) plus 10% foetal bovine serum and 2 mM glutamine are added to each well of a 96-well microtitre plate that is incubated for three days at 37°C. The medium is then replaced with solutions of the active compounds or extracts (diluted in 200 μL of supplemented medium without phenol red) at concentrations 50X the IC50 found in the assay described in Appendix A and the plate is incubated for four days at 37 °C. After this period, 20 μL of AlamarBlueTM is added to each well and the plate is incubated for 4-6 h, after which the absorbance at 570 and 600 nm is measured. Controls with untreated and Bz-treated cells are run in parallel and triplicates are run in the same plate. The results are expressed as the percent difference in the reduction between treated (TC) and untreated cells (UT), using the following equation:
REFERENCES
Copyright © 2010 - Instituto Oswaldo Cruz - Fiocruz The following images related to this document are available:Photo images[oc10041f1.jpg] |
| |||||||||

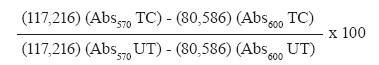
{kind=link}