 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
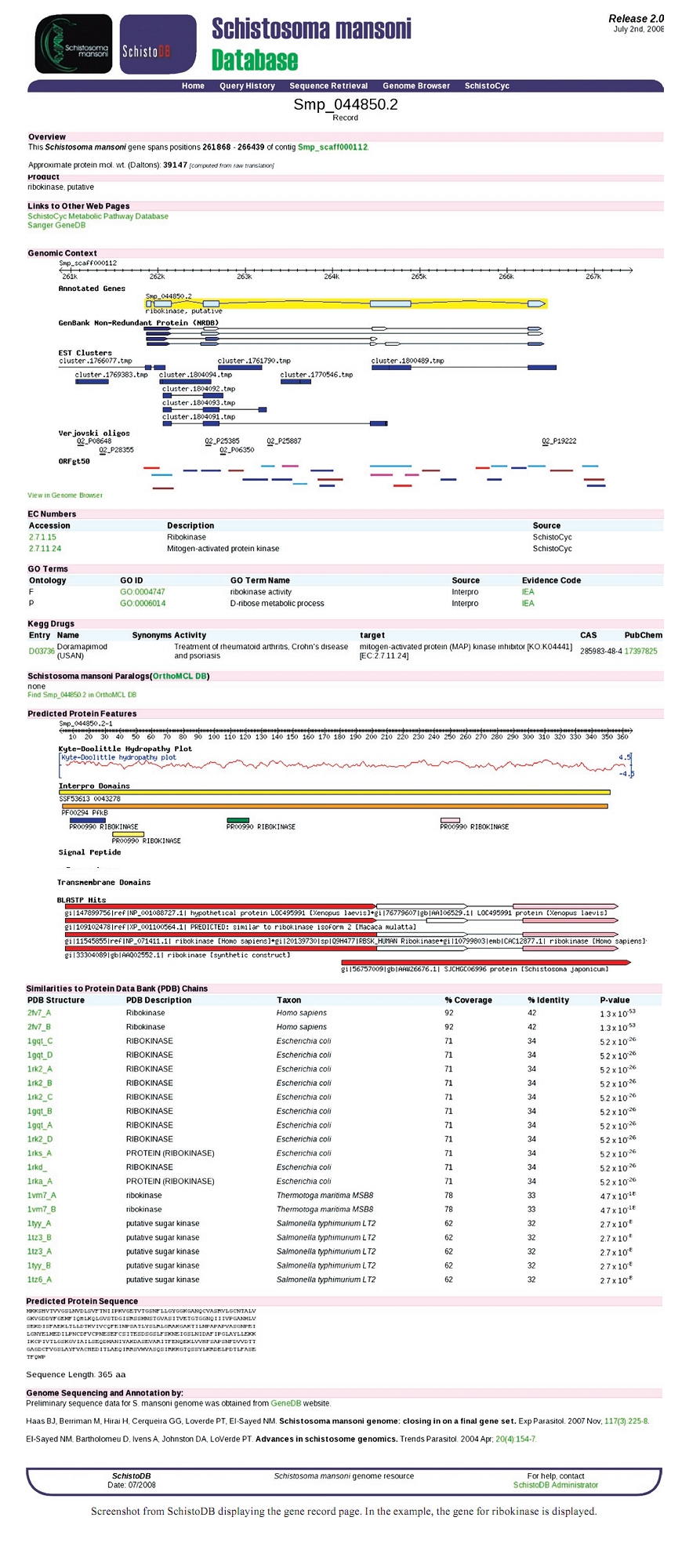
Memórias do Instituto Oswaldo Cruz, Vol. 105, No. 4, 2010, pp. 367-569 The contributions of the Genome Project to the study of schistosomiasis Adhemar Zerlotini; Guilherme Oliveira+ Instituto de Pesquisa René Rachou-Fiocruz, Laboratório de Parasitologia Celular e Molecular, Centro de Excelência em Bioinformática, Av. Augusto de Lima 1715, 30190-002 Belo Horizonte, MG, Brasil + Corresponding author: oliveira@cpqrr.fiocruz.br Received 8 January 2009 Financial support: Fogarty International Center (5D43TW007012-04), FAPEMIG (CBB-1181/0, REDE-186/08, 5323-4.01/07) Code Number: oc10064 ABSTRACT In this paper we review the impact that the availability of the Schistosoma mansoni genome sequence and annotation has had on schistosomiasis research. Easy access to the genomic information is important and several types of data are currently being integrated, such as proteomics, microarray and polymorphic loci. Access to the genome annotation and powerful means of extracting information are major resources to the research community. Key words: Schistosoma mansoni - genome - database - data integration Genome sequencing technologies have considerably expanded our range of tools for experimental and theoretical approaches in the quest for understanding the molecular aspects of schistosomiasis and the design of new control tools. The Schistosoma mansoni genome sequence contains over 360 million base pairs divided into seven pairs of autosomes and one pair of sex chromosomes (female = ZW, male = ZZ) (Berriman et al. 2009). The Wellcome Trust Sanger Institute and an international group of researchers have provided the genome sequencing assembly and annotation (Berriman et al. 2009). The latest draft version of the assembly (Release 4.0) is available online as contigs (50,376) or supercontigs/scaffolds (19,022). Almost half of the genome (45%) was found to be composed of repetitive elements. Both ab initio and evidence based algorithms were used to perform gene prediction and the final automatically annotated sequence includes 11,809 protein-coding gene structures and 13,197 transcripts. It is worth noting that two major Brazilian transcriptome sequencing efforts provided large amounts of expressed sequence tags (EST) (Verjovski-Almeida et al. 2003, Oliveira et al. 2008) that were of critical importance for the identification of the coding regions in the genome. EST data can also be further used for the investigation of transcript variations such as in differential splicing (DeMarco et al. 2006) and alternative polyadenylation (Tian et al. 2007). To infer gene function, several computational analyses were performed using Basic Local Alignment Search Tool (BLAST) (Altschul et al. 1997) for similarity searches, Gene Ontology (Harris et al. 2004) and InterPro (Mulder & Apweiler 2008) for protein domain assignments and limited manual annotation. SchistoDB: S. mansoni genome database - To establish a central repository for S. mansoni genomic data, a database, SchistoDB (Zerlotini et al. 2009) was developed. Similar to other parasite databases with the same architecture (Genomics Unified Schema (Davidson et al. 2001) such as PlasmoDB (Aurrecoechea et al. 2009), ToxoDB (Gajria et al. 2008) and CryptoDB (Heiges et al. 2006), the S. mansoni database provides the community wide access to the latest genome sequence, annotation and other types of data integrated with the genome information. The genome data is structured in a robust relational database coupled with a powerful querying system so that searches can be combined to filter the information based on several criteria. The genome sequences were computationally reanalysed and integrated into a number of public genomic resources. SchistoDB currently provides over 30 different queries and tools for analysis, retrieving or viewing the data. Users can integrate different search results using the "Query History'' page, refining the original query iteratively, until a narrow list of genes of interest is obtained. The data can be downloaded in a flat file format for further analysis and each gene possesses its own record page that contains detailed information of all performed analyses (Supplementary Data). GBrowse genome browser is used to display gene models, EST alignments, BLAST results, protein features etc and facilitates downloading data in various formats. Genomic data analysis - Orthology information provided by the OrthoMCL group (Chen et al. 2006) has been integrated into SchistoDB. In this database orthologous genes from 87 species are clustered based on sequence similarity. The immediate result is the ability to infer protein function through evolutionary relationships, since orthologous genes diverged from a common ancestor owing to speciation events. Additionally orthology information allows us to directly compare S. mansoni genes to other species to narrow a list of candidate drug targets, for example. Using the complete annotated gene set, it is possible to predict the organism's metabolic pathways and gain insight into the physiology of S. mansoni. SchistoDB contains metabolic pathway prediction including approximately 607 enzymatic reactions and 112 pathways that were inferred to occur in the organism based on genome annotation and sequence similarity searches. This information can be used to extend the genome annotation and to compare S. mansoni with other organisms. Several tegumental proteins have been identified as potential vaccine candidates (van Balkom et al. 2005, Braschi et al. 2006b) using proteomic approaches. Such research will benefit from the predicted proteome, not only because it enables the identification of mass fingerprints and peptides, but also because these sequences are computationally characterised to have transmembrane motifs or signal peptides and other types of annotation. Next generation sequencing technologies have become available to S. mansoni research groups, allowing the generation of an extremely large sequence data set in each run. Thus, mapping transcript sequences to the genome, for example, will substantially assist intron/exon boundary validation, thereby improving the gene models and genome assembly. Transcript sequences are also invaluable for alternative splicing, single nucleotide polymorphisms and indel studies. Post-genomic analysis using primarily proteomic and microarray methods is currently being explored by several groups. These experimental approaches, enabled by the genome sequence, have produced essential contributions to a global understanding of how the parasites display sexual differentiation (Waisberg et al. 2008), adapt during development (Jolly et al. 2007) and, for example, how protein expression is compartmentalised (Braschi et al. 2006a). However, these data need to be fully integrated with the genome data to enable the community to make the most use of it. One remaining challenge is identifying the function of the over 40% of unannotated sequences in the genome. Transgenesis and gene silencing by knockout or knockdown experiments will be essential in that process. These technologies remain largely unavailable. However, recent advances were made with the use of RNA interference (Geldhof et al. 2007, Ndegwa et al. 2007). These methods, in combination with the genomic data, will permit a more profound understanding of the biology of schistosomes and undoubtedly the design of new control measures. Genome sequencing and annotation has impacted how molecular research is conducted in schistosomes. Issues related to data sharing and data standards still need to be fully resolved. However, the organisation of the information and the availability of robust querying tools, enabled by a relational genome database such as SchistoDB (http://www.schistodb.net), have provided a framework that provides faster access to the information and empowers groups that are not equipped to conduct the required computational analysis to make use of the information. REFERENCES
Copyright © 2010 - Instituto Oswaldo Cruz - Fiocruz The following images related to this document are available:Photo images[oc10064s1.jpg] |
| |||||||||

{kind=link}