 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Memórias do Instituto Oswaldo Cruz, Vol. 106, No. 2, 2011, pp. 130-138 ARTICLES New insights into trypanosomatid U5 small nuclear ribonucleoproteins Marco Túlio A da SilvaI, II; Daniela L AmbrósioI; Caroline C TrevelinI; Tatiana F WatanabeI; Helen J LaureIII; Lewis J GreeneIII; José C RosaIII; Sandro R ValentiniI; Regina MB CicarelliI, + + Corresponding author: cicarell@fcfar.unesp.br IDepartamento
de Ciências Biológicas, Faculdade de Ciências Farmacêuticas Received 28 July
2010 Code Number: oc11022 ABSTRACT Several protozoan parasites exist in the Trypanosomatidae family, including various agents of human diseases. Multiple lines of evidence suggest that important differences are present between the translational and mRNA processing (trans splicing) systems of trypanosomatids and other eukaryotes. In this context, certain small complexes of RNA and protein, which are named small nuclear ribonucleoproteins (U snRNPs), have an essential role in pre-mRNA processing, mainly during splicing. Even though they are well defined in mammals, snRNPs are still not well characterized in trypanosomatids. This study shows that a U5-15K protein is highly conserved among various trypanosomatid species. Tandem affinity pull-down assays revealed that this protein interacts with a novel U5-102K protein, which suggests the presence of a sub-complex that is potentially involved in the assembly of U4/U6-U5 tri-snRNPs. Functional analyses showed that U5-15K is essential for cell viability and is somehow involved with the trans and cis splicing machinery. Similar tandem affinity experiments with a trypanonosomatid U5-Cwc21 protein led to the purification of four U5 snRNP specific proteins and a Sm core, suggesting U5-Cwc-21 participation in the 35S U5 snRNP particle. Of these proteins, U5-200K was molecularly characterized. U5-200K has conserved domains, such as the DEAD/DEAH box helicase and Sec63 domains and displays a strong interaction with U5 snRNA. Key words: trans splicing - cis splicing - U5 snRNP - U5-Cwc-21 - PTP-Tag - Trypanosoma cruzi - Trypanosoma brucei In trypanosomes, mRNAs are processed by trans splicing, in which a common spliced leader sequence (SL) is acquired at the 5' end of the mRNA to yield a mature transcript (Agabian 1990, Liang et al. 2003). SL trans splicing has been mainly characterized in trypanosomes and nematodes and requires the small nuclear ribonucleoproteins (U snRNPs) U2, U4/U6 and U5, in addition to the SL RNP (Tschudi & Ullu 1990). Intervening sequences have been described in the poly A polymerase (PAP) gene of Trypanosoma brucei and Trypanosoma cruzi and in a U1 small nuclear RNA (snRNA) sequence of T. brucei, which demonstrates that both cis and trans splicing occur in these organisms. Typically, the occurrence of trans splicing is more prevalent (Schnare & Gray 1999, Mair et al. 2000). RNA splicing is carried out by the spliceosome, which consists of the U1, U2, U4, U5 and U6 U snRNPs particles and non-snRNP proteins. The ribonucleoproteins are complexes that consist of small uridine-rich RNAs (U snRNAs) and interact with common Sm proteins and proteins that are specific for each snRNP (Mayer & Floeter-Winter 2005). The detection of the trypanosomatid U5 snRNA was puzzling for many years because the snRNA was not identified in the initial screen for TMG-capped U snRNA. Initially, spliced-leader associated RNA (SLA1) was thought to be the trypanosomatid homologue of U5 because of a sequence that was somewhat related to the invariant U5 loop. The theory that SLA1 was a U5 homologue was disproved because the Leptomonas seymouri homologue did not possess the "invariant" sequence and the "true" U5 was subsequently identified. U5 has several unique properties: it is the shortest snRNA, it lacks several conserved structural domains, including the TMG cap and m7G, and it has a phosphate terminus at the 5' end (Ambrósio et al. 2007). The invariant loop contains an A instead of a C at the second nucleotide position and the loop lacks the characteristic 2'-O-methylation (Liang et al. 2003). The Leptomonas collosoma and the L. seymouri U5 snRNAs are longer than the U5 snRNAs of T. brucei and T. cruzi because they possess a second stem-loop structure (Ambrósio et al. 2007). Analyses of mutations that were introduced into the Sm site of the L. seymouri U5 suggest that U5 snRNA binds to common proteins during a transient cytoplasmic phase before U5 is translocated to the nucleus. This process commonly occurs in mammals but not in yeast (Liang et al. 2003). Although trans splicing is well characterized in humans and yeast, little is known about the specific proteins that mediate trans splicing in trypanosomatids. In human cells, eight U5 snRNP-specific proteins, 220K, 200K, 116K, 102K, 100K, 52K, 40K and 15K, have been identified. Orthologues of the human proteins, PRP8, BRR2, SNU114, PRP6, PRP28, LIN1 or SNU40 and DIB1, respectively, have also been identified in Saccharomyces cerevisiae (Chen et al. 2007). In trypanosomes, the largest protein, PRP8, has been experimentally characterized and appears to function closely with the U5 snRNA (Hodges et al. 1995, Lücke et al. 1997). Genome annotations identified putative orthologues of the U5-116K and U5-15K subunits; recently, two more U5-specific proteins, U5-40K and U5-Cwc21, were identified in T. brucei. Functional analyses demonstrated that U5-Cwc21, a novel U5-specific protein, is essential for cis and trans splicing reactions (Luz Ambrósio et al. 2009). The goal of this study was to identify novel proteins of the trypanosomatid U5 snRNP complex using complex purification techniques, such as PTP-tag. In this study, two different proteins were used as baits, the putative U5-15K and U5-Cwc21. We decided to functionally characterize the trypanosomatid U5-15K protein because the high degree of sequence identity to its orthologue would suggest that the protein participates in cis and/or trans splicing, but previous data demonstrated that the protein only participates in cis splicing. In this study, the results of RNA interference (RNAi) experiments showed that U5-15K is essential for cis and trans splicing reactions in trypanosomatids. Because very little information is available about U5-Cwc21, we decided to use this protein as bait in PTP-tag experiments to identify other related proteins.
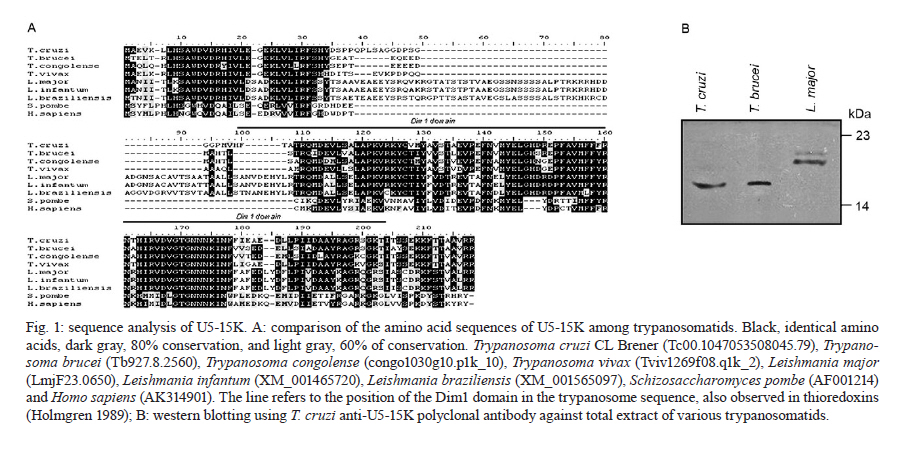
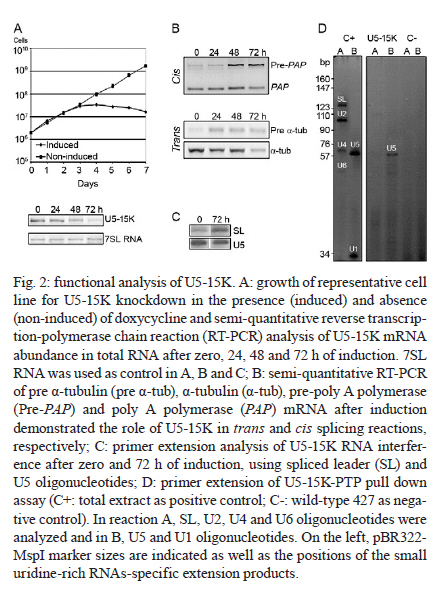
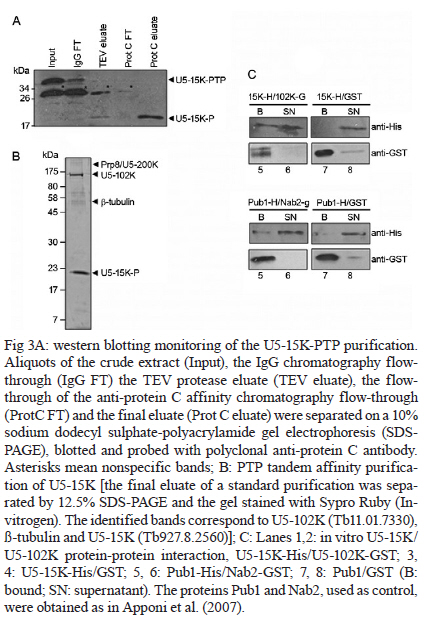
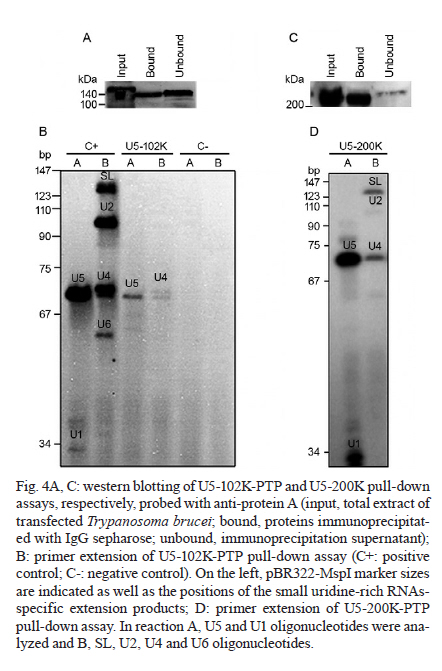
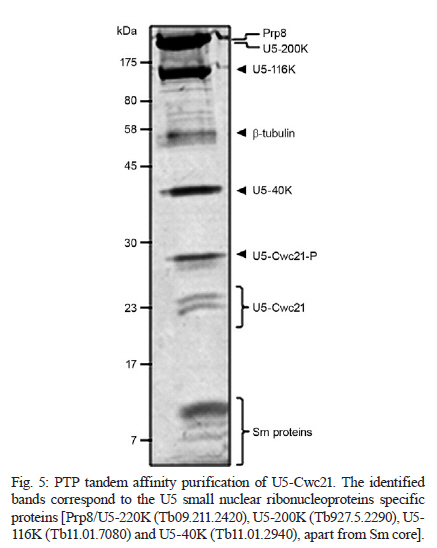
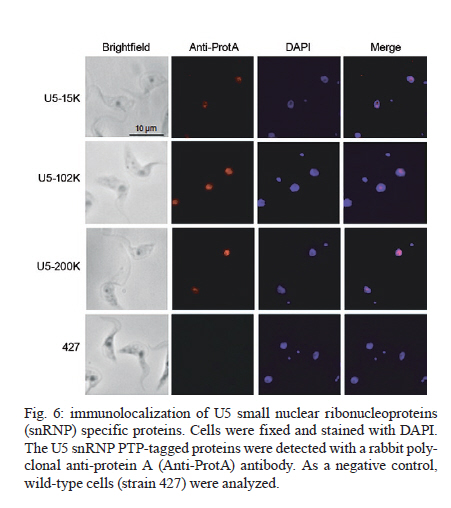
MATERIALS AND METHODS Bioinformatics analyses - Trypanosomatid U5-15K sequences and all of the accession codes were obtained from the GeneDB database (www.genedb.org): T. cruzi CL Brener (Tc00.1047053508045.79), T. brucei (Tb927.8.2560), Trypanosoma congolense (congo1030g10.p1k_10), Trypanosoma vivax (Tviv1269f08.q1k_2) and Leishmania major (LmjF23.0650). Motifs and domains were scanned with the MOTIF SCAN database (http://hits.isb-sib.ch/cgi-bin/PFSCAN). T. cruzi, T. brucei and L. major cultures and DNA extractions - T. cruzi epimastigotes (Y strain) (Silva & Nussenzweig 1953) and L. major promastigotes were grown at 28ºC in liver infusion tryptose medium (Fernandes & Castellani 1966) that was supplemented with 10% foetal bovine serum. T. brucei procyclic cells (strains 427 and 29-13) were grown at 28ºC in SDM-79 (Brun & Schönenberger 1979) medium with the appropriate antibiotics. The parasitic genomic DNA was extracted with the DNAzol reagent (Invitrogen) following the manufacturer's instructions. Expression and purification of recombinant T. cruzi U5-15K - T. cruzi U5-15K, T. brucei U5-15K and T. brucei U5-102K open reading frames (ORFs) were amplified by polymerase chain reaction (PCR) with genomic DNA templates and the following primers: Tc-U5-15K-forward (5'-GGAATTCCATATGGCCGAGGTAAAGCTCC-3'), Tc-U5-15K-reverse (5'-TTGGATCCTTAATTACCGACGCACCGC-3'), Tb-U5-15K-forward (5'-GGATCCATGACGGAGTTGACACGC-3'), Tb-U5-15K-reverse (5'-CTCGAGCTAACGGCGTACA-GCAGC-3'), Tb-U5-102K-forward (5'-GGATCCATGA-GCGCTGCGGCACCTG-3') and Tb-102K-reverse (5'- GCGGCCGTTACAATTTGATCATTTC-3'). The PCR products were sequenced in both directions and cloned into the pGEM-T Easy Vector (Promega). The PCR fragments of T. cruzi and T. brucei U5-15K were sub-cloned into the pET28a expression vector and the PCR fragments of T. brucei U5-102K were sub-cloned into pGEX-4T1 (Amersham Biosciences). Recombinant proteins were expressed in Escherichia coli BL21(DE3) pLysS cells after the induction of protein production with 0.4 mM isopropyl-β-D-thiogalactopyranoside. For purification of the recombinant proteins, cell pellets were resuspended in a lysis buffer (50 mM sodium phosphate pH 8.0, 300 mM NaCl and 10 mM imidazole) and the cells lysed by sonication. Cell debris was removed by centrifugation (20,000 g for 15 min at 4ºC) and the recombinant T. cruzi and T. brucei U5-15K-H6 and U5-102K-GST tagged proteins were isolated from the supernatants by Ni-NTA and glutathione Sepharose 4B affinity chromatography, respectively, in the Äkta FPLC system (Amersham Biosciences) following to the manufacturer's instructions. To obtain U5-15K polyclonal antibodies, approximately 200 μg of purified recombinant T. cruzi U5-15K was inoculated into rabbits and the sera were collected. Pre-immune sera were collected before the rabbits were immunized with the recombinant protein. PTP-tag pull-down assays - For the PTP-tagged T. brucei U5-15K (Tb927.8.2560) and U5-Cwc21 (Tb09.160.2110) constructs, the full ORFs of each gene were amplified with the following primers that contained EagI or ApaI restriction sites: 15K-PTP-F (5'-CGGCCGACGGCGTACAGCAGCTGTTG-3'), Cwc21-PTP-F (5'-TACGGCCGCTCTCCCTTGATACG-CTTCA-3'), 15K-PTP-R (5'-GGGCCCATGACGGAGTTGACACGC-3') and Cwc21-PTP-R (5'-TAGGGCCC-CTGGAGGCGTTGCGGTCGGC-3'). The ORFs were then inserted in-frame into the pC-PTP-NEO vector upstream of the PTP tag sequence using the EagI and ApaI restriction sites. The resulting constructs were subsequently linearized with the BbsI or PshAI restriction enzymes. For the U5-102K (Tb11.01.7330) and U5-200K (Tb927.5.2290) proteins, the ORFs were partially cloned into the pC-PTP-NEO vector, nucleotides +2351 to +3027 and +6122 to +6711, respectively. The following primers were used to amplify the ORFs: 102K-PTP-F (5'-CGGCCGCAATTTGATCATTTCCACGA-3'), 102K-PTP-R (5'-GGGCCCTGCTTCAGCAACTTCCT-AAG-3'), 200K-PTP-F (5'-CGGCCGTCCCGCTGCAA-AGGTTAC-3') and 200K-PTP-R (5'-GGGCCCTCCAAGAGCTGTGCGATG-3'). The resulting constructs were linearized with AflII or StuI. To generate cell lines that expressed the PTP-tagged proteins, 10 μg of the linearized PTP vectors were transfected into T. brucei procyclic cells (strain 427) by electroporation (Lee 1996). Transfected cells were selected with limiting dilutions with 40 μg/mL of G418 (Sigma). The correct integrations of the constructs were confirmed by PCR and PTP-tagged protein expression was analyzed by immunoblotting with an anti-protein A antibody. In the PTP-tag pull-down assays, 100 μL of cell extracts from stably transfected cells were incubated at 4ºC with 30 μL of packed IgG Sepharose 6 Fast Flow beads (GE Healthcare) that had been previously equilibrated with PA-150 buffer (150 mM NaCl, 20 mM Tris-HCl, pH 7.7, 3 mM MgCl2, 0.5 mM DTT, and 0.1% Tween 20). After washing the beads with the same buffer, total RNA was extracted from the beads with TRIzol reagent (Invitrogen) and then resuspended in 40 μL of deionized water. The following 32P-end-labeled DNA oligonucleotides were used in primer extension reactions: SL PE (52 -CGACCCCACCTTCCAGATTC-32 ), U2 PE (52 -ACAGGCAACAGTTTTGATCC-32 ), U4 PE (52 -TACCGGATATAGTATTGCAC-32 ), U6 PE (52 -GGGAGAGTGCTAATCTTCTC-32 ), U1 PE (52 -AGC-ACGGCGCTTTCGTGATG-32 ) and U5 PE (52 -CCG-CTCGAGGACACCCCAAAGTTT-32 ). Primer extension reactions were performed with 10 μL of total RNA and the SuperScript II reverse transcriptase (Invitrogen) following the manufacturer's instructions. Extension products were separated by electrophoresis on 8% polyacrylamide gels with 8 M urea and were then analyzed with a Typhon Trio-Variable Mode Imager (GE Healthcare). T. brucei wild-type extracts were used as positive controls. The same PTP tag pull-down protocol was used to confirm the ability of U5-15K PTP-tagged proteins to interact with the IgG Sepharose. Supernatants and resins from the pull-down experiments were analyzed by immunoblotting using an anti-protein A antibody. PTP-tag purifications were performed as described by Schimanski et al. (2005). Proteins that were released were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and stained with Sypro Ruby (Invitrogen) and Coomassie blue. Gel in situ trypsin digestions and mass spectrometry analyses - The samples were separated by 12.5% SDS-PAGE and stained with Coomassie blue. Gel bands that corresponded to the tagged proteins were excised and de-stained with 50% acetonitrile/0.1 M ammonium bicarbonate pH 8.0. Gels were dehydrated with neat acetonitrile and dried in a Speed-Vac for 30 min. The gels were then swollen with 0.5 μg of modified trypsin (Promega) in 20 μL of 0.1 M ammonium bicarbonate pH 8.0. Next, the gels were covered with more than 100 μL of 0.1 M ammonium bicarbonate pH 8.0 for 22 h at 37ºC. Trypsin hydrolysis was stopped with 5 μL of neat formic acid. The tryptic peptides were extracted and desalted in a microtip that was filled with POROS 50 R2 (PerSeptive Biosystems), which was previously equilibrated in 0.2% formic acid. The peptides were eluted with 60% methanol/5% formic acid for subsequent mass spectrometry analyses. Eluted samples were diluted in a matrix solution (5 mg/mL of alfa-cyano-4-hydroxicinnamic acid, Sigma, with 50% acetonitrile/0.1% trifluoroacetic acid). Two or 3 μL of the samples were then spotted on Maldi plates and allowed to air dry. Mass spectrometry analysis was performed using a MALDI-TOF/TOF (Axima Performance, Kratos-Shimadzu) and the MS/MS spectra of tryptic peptides were directly analyzed by using a Mascot program search against the NCBInr and local trypanomatid databases. In vitro binding assays - To confirm protein-protein interactions in vitro, binding assays were performed as described by Apponi et al. (2007), with some modifications. One microgram of GST or GST-fused protein was bound to 30 μL of glutathione-Sepharose (GE) in phosphate-buffered saline for 30 min at 4ºC. After three washes with 500 μL of phosphate-buffered saline, 400 ng of a His6-tagged protein was added to 500 μL of buffer A [20 mM Tris-HCl (pH 8.0), 0.5% Triton X-100 and 50 mM NaCl] and the mixture was incubated for 1 h at 4ºC. The mixture was then washed three times with the same buffer and unbound and bound fractions were analyzed by immunoblotting using anti-GST (GE) and anti-His (Sigma) antibodies. RNAi experiments - For T. brucei U5-15K RNAi, stem-loop constructs were generated following a published cloning strategy (Shi et al. 2000). The constructs contained the sequence from positions +65-468, which was amplified with the following primers: U5-15K-RNAi forward (5'-AAGCTTACGCGTAGAAACTTGTACTTATCCGC-3') and U5-15K-RNAi reverse (5'-TCTAGAACGGCGTACAGCAGCTGTTGT-3'). The amplified products were cloned into the pLEW100 vector. To generate the RNAi cell lines, T. brucei strain 29-13 cells were transfected and clonal populations of transfected cells were isolated with limiting dilutions and selected with 2.5 μg/mL of phleomycin (Sigma). To monitor the growth of RNAi cells, dsRNA synthesis was induced with 2 μg/mL of doxycycline (Sigma). The cells were counted daily and diluted to a concentration of 2 × 106 cells/mL. Reverse transcription (RT)-PCR and primer extension experiments were used to monitor the silencing and splicing defects. To determine the relative abundance of specific transcripts in RNAi cells, total RNA preparations from whole cell lysates were analyzed by semi-quantitative RT-PCR assays. For each PCR, the cycle numbers of the linear amplification range were determined. Oligonucleotides that were used in cis and trans splicing experiments to amplify PAP, pre-α tubulin, α tubulin and 7SL RNA were previously described by Luz Ambrósio et al. (2009). U5-15K specific transcripts were amplified with U5-15K forward and AUAP primers (Invitrogen). The RNA preparation and primer extension protocols mentioned above were also used to analyze the levels of U snRNAs in doxycycline-induced RNAi cells. Immunolocalizations - Immunofluorescence assays were performed as described by Luz Ambrósio et al. (2009). Images with identical exposure settings were taken with TM Leitz DM RXZ microscope (Leica) and analyzed with Motic Images Advanced 3.2 software (Motic). RESULTS Analyses of trypanosomatid U5-15K sequences - The U5-15K protein likely plays an important role in the structure and function of the U5 snRNP, which is corroborated by its high degree of sequence similarity to other trypanosomatid U5-15K orthologues. T. cruzi U5-15K exhibits 47% and 46% amino acid identity to human (AK314901) and yeast (AY558054.1) orthologues, respectively. The T. cruzi U5-15K ORF, which is 498 nucleotides, encodes a protein that is 162 amino acids and has an estimated molecular weight of 18.062 kDa. The conservation of U5-15K sequences among various trypanosomes ranges from 42-72% identity at the nucleotide level and from 36-76% identity at the amino acid level (Fig. 1A). Unexpectedly, the ORF of L. major U5-15K is 655 nucleotides, which is larger than the other U5-15K ORFs and is 42% identical at the nucleotide level and 36% identical at the amino acid level to T. cruzi U5-15K. Similar results were also observed with the Leishmania infantum and Leishmania braziliensis U5-15K ORFs. Leishmania species have an additional insertion between amino acid residues 48 and 92. The 44 amino acid insertion is responsible for the 8.3 kDa size increase in the Leishmania U5-15Ks relative to the Trypanosome species (Fig. 1A, B). Western blots, using an anti-T. cruzi U5-15K polyclonal antibody, of the total (data not shown) and nuclear extracts of T. cruzi, T. brucei and L. major showed the presence of a native 17 kDa protein in the first two species and a larger protein in L. major (Fig. 1 B). The presence of a 21 kDa band (lower band) confirmed the existence of an insertion in the Leishmania protein. The biological significance of this insertion has not yet been explored. The upper band that is present in the Leishmania lane is probably background from a nonspecific interaction of the T. cruzi antibody. Molecular and functional data confirm the participation of U5-15K in splicing processes - The involvement of U5-15K in splicing reactions was demonstrated by RNAi. Growth curves of the doxycycline-induced and uninduced cells showed that after three days of induction, the induced cells displayed declining growth rates, which continued until the seventh day (Fig. 2A). Twenty-four hours after induction, the mRNA levels of U5-15K steadily declined and the maximum inhibition was reached 72 h after induction (Fig. 2A). The levels of the control 7SL RNA, which is RNA that is present in the signal recognition particle and is essential for protein translocation across the endoplasmic reticulum, remained constant during the course of the experiment. These data suggest that the U5-15K gene is essential, and its absence significantly reduces the normal growth rate of the parasite. To demonstrate the involvement of U5-15K in the trans and cis splicing reactions, we performed RT-PCR reactions using the same conditions as those described by Luz Ambrósio et al. (2009). The tubulin mRNA primers amplified unprocessed mRNA and mRNA that had been processed by trans splicing. Semi-quantitative RT-PCR showed that tubulin mRNA processing by trans splicing was inhibited when RNAi cells were induced (Fig. 2B, pre-α and α-tub). The pre-α tub RNA accumulated 72 h after induction, whereas the processed α-tubulin mRNA was strongly reduced at the same timepoint. The participation of U5-15K in cis splicing was confirmed by RT-PCR using primers that were previously described by Mair et al. (2000) and flanked the intron of PAP, whose mRNA is processed by cis splicing in trypanosomes. During the course of the induction, unprocessed PAP transcripts accumulated (approximately 780 bp band) (Fig. 2B) and the mature transcript level was discretely reduced (130 bp band) (Fig. 2B), especially 72 h after induction. Similar to the observations of mammals and yeast (Simeoni & Divita 2007), these data confirm the participation of U5-15K in the cis splicing reaction and suggest that the U5 snRNP is essential in cis and trans splicing reactions in trypanosomatids. The level of U5 snRNA did not change during the induction. This result indicates that even though the protein was absent, the structural RNA was stable, which was demonstrated by primer extension experiments (Fig. 2C). A discrete accumulation of SL RNA was observed (Fig. 2C), which confirms that the trans splicing reaction was inhibited and suggests that U5-15K may be involved in the first step of the trans splicing reaction. To analyze the proteins that interacted with U5-15K, we established the U5-15K-PTP T. brucei cell line. The protein that was expressed in this cell line displayed a band of 34 kDa (17 kDa of U5-15K + 19 kDa of PTP-tag), which indicates that this protein correctly fused to the tag. T. brucei U5-15K-positive clones were used to demonstrate that an interaction occurred with structural RNA and primer extension assays showed that U5 snRNA either directly or indirectly associated with U5-15K (Fig. 2D). These data confirm the involvement of U5-15K in the T. brucei U5 snRNP complex. Furthermore, because a high level of conservation was observed in the splicing reaction, other trypanosomatid U5-15K proteins likely behave similarly. Tandem affinity purification with U5-15K reveals a novel protein-protein interaction between trypanosomatid splicing proteins - The clone that expressed the appropriate PTP-tag, which was confirmed by western blots and PCR, was used for the PTP-tag purification as described by Schimanski et al. (2005). The steps of PTP-tag purification were monitored by western blot analyses with an antibody that was directed against protein C in the tag (Fig. 3A). The input U5-15K-PTP protein was approximately 34 kDa. In the next step of the purification process, a considerable portion of U5-15K-PTP interacted with the IgG chromatography flow-through (IgG FT). The U5-15K protein that was fused with only protein C (U5-15K-P) was released after being cleaved by the TEV protease and displayed a molecular weight of approximately 22 kDa (TEV eluate). U5-15K-P enrichment occurred after the protein interacted with the anti-protein C resin and was eluted (Prot C eluate). The protein band that is marked in Fig. 3A represents a nonspecific band. The nonspecific band did not interfere with the experiments because it was completely eliminated during the purification process. After the final eluate was separated by SDS-PAGE, two major bands were observed (Fig. 3B). The 22 kDa protein band corresponded to the U5-15K protein that was fused with protein C and was identified in the T. brucei genome database as Tb927.8.2560. The higher molecular weight band was identified as a conserved hypothetical protein, Tb11.01.7330. PTP-tag experiments that were previously performed by Luz Ambrósio et al. (2009) with T. brucei SmD1 identified a hypothetical protein in T. brucei, Tb11.01.7330, which the authors classified as U5-102K. However, Palfi et al. (2009) did not detect this protein in more recent experiments with SmB-PTP. In vitro experiments were performed to determine if an interaction occurs between U5-15K and U5-102K without intermediate ligands, such as RNA and other factors. U5-15K and U5-102K were expressed and purified with different protein tags (U5-15K-His and U5-102K-GST). In Fig. 3C, we show that U5-15K-His binds directly to U5-102K-GST (Lane 1). The GST tag alone does not bind (Lane 3), which confirms that an interaction exists between these two members. The primer extension assays in Fig. 2D show that small amounts of U5 snRNAs are co-purified in U5-15K-PTP, which is likely the result of U5-15K participating in another complex. Small amounts of Prp8/U5-200K purified reinforce the possibility of the other complex purification, such as 20S U5 snRNP (Fig. 3B). Molecular data confirms that U5-102K is a U5 snRNP specific protein - The ORF of U5-102K is highly conserved among Trypanosome species and contains 2985 nucleotides in T. cruzi and 3030 nucleotides in T. brucei. In L. major, the U5-102K ORF is only a partial sequence. The identities of the amino acid sequences range from 59% in T. cruzi and T. brucei to 42% in T. cruzi and T. vivax (data not shown). Bioinformatics analysis with the T. brucei U5-102K sequence as template revealed that one HAT motif was present between positions 310 and 341 and a tetratricopeptide (TPR) was present between positions 847 and 880. However, the identities of the motifs were low. The yeast human Prp6 protein (hPrp6) contains an N-terminal domain that is followed by 13 HAT (Half-A-TPR) repeats and one TPR motif. The HAT motif is found in several RNA-binding proteins (Preker & Keller 1998) and it shares structural and sequence similarities to the TPR repeats, which mediate protein-protein interactions (Liu et al. 2006). To confirm that U5-102K was involved in the U5 snRNP complex, a cell line that expressed PTP-tagged U5-102K was obtained. In western blots that used an anti-protein A antibody (Input, Fig. 4A), a band of approximately 140 kDa was observed, which confirmed the presence of PTP-tagged U5-102K (109 kDa of U5-102K + 19 kDa PTP-tag). Extracts from transfected T. brucei cultures were tested for the ability of the protein to bind to the IgG-Sepharose resin (Fig. 4A). Primer extension experiments that involved immunoprecipitating U5-102K with IgG-Sepharose indicated the presence of U5 snRNA, which suggests that U5-102K probably participates in the U5 snRNP complex (Fig. 4B). Small amounts of U4 snRNA were also obtained, which indicates that the tri-snRNP U5/U4/U6 may have also been purified, albeit in small amounts. Notably, the association of the tri-snRNP U5/U4/U6 complex occurs before the final assembly of the B complex (Liu et al. 2006). U5-Cwc21 forms a stable complex with Sm and four U5 snRNP specific proteins in the 35S complex - To identify the proteins that interact with T. brucei U5-Cwc21, we established an U5-Cwc21-PTP cell line and confirmed protein interactions using IgG Sepharose and western blot analyses (data not shown). PTP-tag purifications were performed following the standard protocol of Schimanski et al. (2005). The purification steps were monitored by western blot analysis using an anti-protein C antibody as previously described in this study for the U5-15K-PTP experiments (data not shown). U5-Cwc21-PTP purifications (Fig. 5) and mass spectrometry analyzes revealed four U5-snRNP specific proteins in addition to the Sm core (Palfi et al. 2000): Prp8 (Tb09.211.2420) (Lücke et al. 1997), U5-200K (Tb927.5.2290), U5-116K (Tb11.01.7080) and U5-40K (Tb11.01.2940) (Luz Ambrósio et al. 2009). A doublet was also observed in the purification and was later identified as native Cwc-21 (without the tag), which suggests the protein forms dimers in vivo. The presence of the doublet that is related to native Cwc-21 (Fig. 5) suggests that different isoforms of the protein exist or that post-translational modifications occur. Similarly, western blot analyzes with an anti-Cwc-21 polyclonal antibody also detected two bands in total extracts from T. brucei and T. cruzi (data not shown). High U5 snRNA interaction levels confirm that U5-200K is a U5 snRNP specific protein - Among all of the proteins that were identified by mass spectrometry in U5-Cwc21-PTP assays, U5-200K was the only protein that was not characterized as a U5 snRNP specific protein. However, two independent studies that were conducted with bioinformatics tools after the conclusion of this work described U5-200K as a U5 snRNP specific protein (Günzl 2010, Tkacz et al. 2010). To determine the structural RNA that binds to U5-200K, a T. brucei U5-200K-PTP cell line was established. The binding capacities of proteins that were produced in this cell line were tested with an IgG FT (Fig. 4). Primer extension experiments demonstrated that U5-200K-PTP co-purified with all U snRNAs, including SL RNA. The U5 snRNA interaction level was higher (Fig. 4) than the other snRNAs in comparison to the positive control (Fig. 4). These data suggest that U5-200K participates in complexes that are directly associated with U5 snRNA. Furthermore, the presence of the other RNAs shows that U5-200K probably co-purifies various particles that are formed by different U snRNPs, such as the B, B*, C and 35S U5 snRNP complexes. Unexpectedly, U2 snRNA levels were low. The presence of U1 snRNA and SL RNA suggest a role for U5-200K in cis and trans splicing reactions (Fig. 4). T. brucei U5-200K has an estimated molecular weight of 268 kDa and an isoeletric point of 5.75. The DEAD/DEAH box helicase domain was identified between positions 404 and 584 in T. brucei. In other organisms, this domain is involved in various aspects of RNA metabolism, which include nuclear transcription, pre-mRNA splicing, ribosome biogenesis, nucleo-cytoplasmic transport, translation, RNA decay and organelle gene expression. In T. brucei U5-200K, a second DEAD/DEAH box helicase domain was identified between amino acids positions 1273 and 1315. Whereas the motifs in the first helicase-like domain of S. cerevisiae Brr2 (yBrr2) are critical for ATPase activity, U4/U6 unwinding, and cell viability, the motifs in the second helicase domain can be disrupted with little consequence (Zhang et al. 2009). Trypanosomatid U5-200K exhibits two Sec63 domains at amino acids positions 907-1060 and 1116-1220, which are different than the locations of the same domains in yeast Brr2. Both of these domains are located in a central region of the trypanosome orthologue, but the yeast orthologue has one domain in a central region and the other domain in the N-terminal region. Recent data obtained from yeast suggest that the Sec63 unit of the Brr2 N-terminal helicase cassette is an integral component of the active site and could allow Brr2 to unwind the extended U4/U6 duplex during spliceosome catalytic activation (Pena et al. 2009). The trypanosome U5 snRNP proteins are localized in the nucleus - The U5-15K, -102K and -200K PTP-tagged proteins were visualized in cells with an anti-protein A antibody (Fig. 6). The proteins appeared as speckles that were concentrated in the nucleus, which is consistent with proteins that are involved in splicing processes. Luz Ambrósio et al. (2009) also observed T. brucei Cwc-21 in the nucleus. DISCUSSION The spliceosomal U5-15K protein is a U5-snRNP specific protein that has been described in human (U5-15K) and yeast cells (Dib1p). The role of Dib1p in pre-mRNA splicing has been shown by genetically depleting the protein in vivo in the S. cerevisiae system (Reuter et al. 1999). In Schizosaccharomyces pombe, the orthologue of U5-15K, Dim1p, is required for efficient pre-mRNA splicing and has an essential function in mediating the export of certain mRNAs from the nucleus (Carnahan et al. 2005). The data show that U5-15K is highly conserved from kinetoplastids to human and functions in cis and trans splicing reactions, which suggests that the function of this protein emerged early in the evolution of mRNA processing. Co-purifications of U5-15K with U5-102K, which were confirmed by in vitro and in vivo experiments, suggest the possibility of a sub-complex that is composed solely of these two strongly interacting proteins. U5-15K and U5-102K may be present as a stable complex in the nucleus and the complex may be incorporated into the 35S U5 snRNP complex to restore the 20S U5 snRNP complex before its association with the U4/U6-U5 tri-snRNP complex. These data are supported by the fact that Dib1 interacts with the N-terminus of Prp6 (Liu et al. 2006). In addition, a recent study in yeast showed that mutations in the N-terminus of Prp6 prevented the incorporation of the protein into the spliceosome, which ultimately lead to an accumulation of pre-catalytic complexes (Lützelberger et al. 2010). Therefore, it is possible that changes in the interactions between Prp6 and Dib1 prevent the incorporation of the tri-snRNP complex, which blocks the completion of the catalytic complex. However, further studies with U5-15K and U5-102K are required to confirm the function of the sub-complex that is formed by these two proteins. Structural models have demonstrated that U5-15K forms a binary complex with U5-52K, which is mediated by a GYF-domain, in human cells (Nielsen et al. 2007). In addition, U5-15K is also able to bind to another U5-specific protein that is named U5-102K (Liu et al. 2006). Yeast two-hybrid screens and pull-down assays revealed that the 52K protein interacts with the U5-specific 102K and 15K proteins, suggesting that these interactions are responsible for the integrations of these proteins into the U5 particle. The interaction of 52K with a tri-snRNP bridging protein, U5-102K, and its absence from the tri-snRNP complex suggests that the protein might function in tri-snRNP assembly (Laggerbauer et al. 2005). As U5-52K is absent from trypanosomes, the possibility exists that the U5-15K/U5-102K sub-complex is necessary for tri-snRNP assembly. Mutational experiments with the yeast Prp6 protein and previous biochemical analyses and RNAi-mediated knockdowns of the human hPrp6 suggest that Prp6 plays a crucial role in establishing a connection between the U5 and U4/U6 snRNPs (Schaffert et al. 2004). In trypanosomes, U5-102K might also act in the U4/U6-U5 tri-snRNP complex because U4 was co-purified with U5 snRNA. This observation is corroborated by the presence of Prp31 in trypanosomatids and functional data suggest that T. brucei Prp31 stabilizes the U4/U6-U5 tri-snRNP complex and possibly participates in the formation of the complex (Liang et al. 2006). However, additional interaction experiments are required to confirm a possible physical interaction between Prp31 and U5-102K in trypanosomes. Makarov et al. (2002) identified the proteins that were present in the 35S U5 snRNP particle in HeLa cells, which included U5 snRNP specific proteins (220K, 200K, 116K and 40K) and other spliceosomal proteins that were mainly from the hPrp9 complex. Moreover, proteins from the Prp19 complex, some snRNP proteins and other spliceosomal proteins were purified with TAP-tagged Cwc21 from yeast, but only Prp8, Prp6 and Brr2 appeared to be U5 snRNP proteins (Grainger et al. 2009, Khanna et al. 2009). The T. brucei U5-Cwc21 description suggests that this protein belongs to the Prp19 complex or the 35S U5 snRNP complex (Luz Ambrósio et al. 2009). Although homologues of hPrp9, CDC5, hSyf1, hIsy1 and Syf3, which are Prp19 complex proteins, have been identified in trypanosomes, proteins that are related to the Prp19 complex were not co-purified with U5-Cwc21-PTP (Günzl 2010). The presence of Prp8, 200K, 116K and 40K in Cwc-21 purifications strongly suggests that T. brucei U5-Cwc-21 is a subunit of the 35S U5 snRNP particle. However, the absence of prp19 proteins does not exclude the possibility of an interaction between Prp19 and U5 snRNP complexes. ACKNOWLEDGEMENTS Trypanosoma brucei strains (427 and 29-13) were a kind gift from Dr Otávio H Thiemann, Instituto de Física de São Carlos, Universidade de São Paulo. PTP and RNAi vectors were gently provided to Dr Arthur Günzl, University of Connecticut Health Center, Farmington, USA. REFERENCES
Copyright © 2011 - Instituto Oswaldo Cruz - Fiocruz The following images related to this document are available:Photo images[oc11022f2.jpg] [oc11022f1.jpg] [oc11022f5.jpg] [oc11022f4.jpg] [oc11022f3.jpg] [oc11022f6.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}