 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Memórias do Instituto Oswaldo Cruz, Vol. 106, No. 2, 2011, pp. 177-181 ARTICLES The impact of the nelfinavir resistance-conferring mutation D30N on the susceptibility of HIV-1 subtype B to other protease inhibitors André FA SantosI; Marcelo A SoaresI, II, + ILaboratório
de Virologia Humana, Departamento de Genética, Centro de Ciências
da Saúde, Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ,
Brasil + Corresponding author: masoares@biologia.ufrj.br Received 5 August
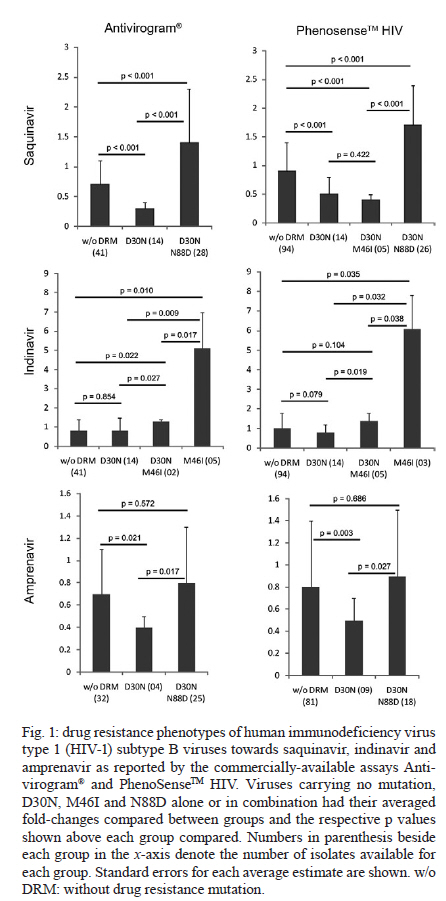
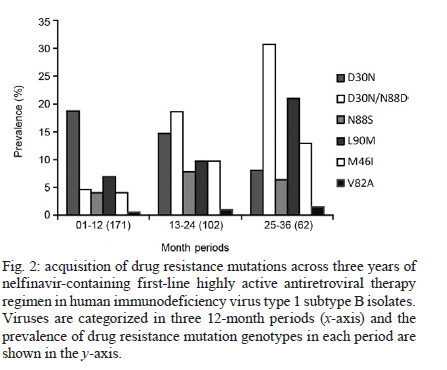
2010 Code Number: oc11029 ABSTRACT The human immunodeficiency virus type 1 (HIV-1) protease mutation D30N is exclusively selected by the protease inhibitor (PI) nelfinavir and confers resistance to this drug. We demonstrate that D30N increases the susceptibility to saquinavir (SQV) and amprenavir in HIV-1 subtype B isolates and that the N88D mutation in a D30N background neutralizes this effect. D30N also suppresses indinavir (IDV) resistance caused by the M46I mutation. Interestingly, in patients with viruses originally containing the D30N mutation who were treated with IDV or SQV, the virus either reversed this mutation or acquired N88D, suggesting an antagonistic effect of D30N upon exposure to these PIs. These findings can improve direct salvage drug treatment in resource limited countries where subtype B is epidemiologically important and extend the value of first and second line PIs in these populations. Key words: D30N - hypersusceptibility - phenotyping - drug resistance - protease inhibitors - N88D The human immunodeficiency virus type 1 (HIV-1) of subtype B (HIV-1B) was responsible for only ~10% of new infections in 2004, but it predominates in developed countries including the United States, Canada, western European countries, Japan and Australia (Hemelaar et al. 2006). In Brazil, as is the general trend in Latin America, the predominant HIV-1 subtype is B with a prevalence of 66-72% according to the most recent studies (Inocencio et al. 2009, Sprinz et al. 2009). Thus, HIV-1B is the most studied subtype in the contexts of drug design, highly active antiretroviral therapy (HAART) regimen optimization, drug resistance mutation (DRM) acquisition and drug phenotypic susceptibility. Consequently, HIV-1B is the model for studying drug resistance. HIV-1 is able to acquire DRMs for all commercially available antiretroviral drugs (Johnson et al. 2009). Currently, the emergence of DRMs is the main cause of therapeutic failure in clinical HIV/acquired immunodeficiency syndrome (AIDS) patients, with a subsequent viral load increase and loss of CD4+ T-cells and, consequently, progression to AIDS (Clavel & Hance 2004). HIV-1 hypersusceptibility (HS) to a given drug occurs when a polymorphism in a viral gene increases the susceptibility to that drug compared to a wild-type virus. There are some reports of DRMs of one drug causing HS to other drugs. The most well-known case is M184V in HIV-1 reverse transcriptase, which confers resistance to 3TC, but also increases the susceptibility to thymidine analogues including zidovudine (AZT) and stavudine (d4T) (Larder et al. 1995). In the protease (PR) region, the N88S mutation causes resistance to nelfinavir (NFV) and atazanavir (ATV), but HS to amprenavir (APV) (Ziermann et al. 2000, Mitsuya et al. 2006). I50L increases ATV resistance and antagonistically increases susceptibility to other protease inhibitors (PIs) (Ziermann et al. 2000). According to the International AIDS Society panel of HIV-1 DRM, the D30N mutation is considered a major mutation for NFV and it confers a significant loss of susceptibility to this drug (Johnson et al. 2009). In addition, the D30N and N88D mutations are exclusively acquired upon NFV treatment and do not have major or compensatory effects for other PIs. However, the resistance profiles to NFV and saquinavir boosted with ritonavir (SQV/r) are incomplete, despite being the first PIs released for clinical use. Moreover, N88D is strongly correlated with D30N and its presence allows the acquisition of L90M in a D30N background (Sugiura et al. 2002, Mitsuya et al. 2006). More recently, Rhee et al. (2006) showed through bioinformatic analyses that D30N increases viral drug susceptibility to APV, indinavir (IDV), lopinavir (LPV) and SQV/r. However, the clinical impact of this phenomenon has not yet been investigated. In the present study, we evaluated the phenotypic role of D30N and D30N/N88D in clinical HIV-1 subtype B isolates, in addition to the acquisition of these mutations over NFV exposure time and their persistence to posterior SQV/r or IDV salvage therapies. PATIENTS, MATERIALS AND METHODS Sequences from 184 clinical isolates of HIV-1 subtype B with drug phenotypic susceptibility determined by Antivirogram® (Virco, Belgium) (Hertogs et al. 1998) and sequences from 182 isolates with susceptibility determined by PhenoSenseTM HIV (Monogram, United States) (Kellam & Larder 1994) were retrieved from the Stanford HIV Drug Resistance Database (Shafer et al. 2000). These viral sequences contained known DRMs selected by NFV (D30N, M46I, V82A, I84V, N88D and L90M) (Johnson et al. 2009) and were grouped according to the phenotypic assay used and the DRM genotypes (single or double mutants). Fold-change (FC), i.e., the ratio of the drug concentration necessary to inhibit 50% of the clinical virus (IC50) and the IC50 from HIV-1 subtype B infectious clone HXB2 (wild-type control) was calculated for each drug. FC values were averaged among samples from each group. The PIs fosamprenavir, ATV, IDV, LPV, NFV and SQV were evaluated. Differences between groups were statistically analyzed with a two-tailed Student's t test and p values < 0.05 were considered significant. To evaluate the emergence of resistance across time of exposure, we obtained genotypes of subtype B viral isolates from the Stanford Database coupled with complete history treatment, including the number of regimens, regimen composition and time of treatment. In this study, we only analyzed patients undergoing their first NFV-containing HAART drug regimen. Patients with historical use of mono and/or dual therapy were excluded. Isolates were divided into annual periods of therapy exposure and the proportion of each major DRM was calculated for each time period. We also retrieved viral isolates from clinical patients containing D30N or D30N/N88D mutations with NFV failure treatment from the Stanford Database. These patients had an additional genotyping time-point: after they changed treatment to SQV/r or IDV. We evaluated which DRMs emerged and/or reversed during the PI treatment switch. RESULTS The two commercial phenotyping assays did not show significant differences in the average FC values (Table). There were only five (6%) susceptible genotypes according to Antivirogram® that showed resistance according to PhenoSenseTM HIV; the opposite was observed in only three cases (4%). In most of these cases, the low number of isolates with a definite genotype may explain the differences. In the remaining 70 comparisons (90%), both phenotyping assays were concordant in the susceptibility status. The D30N mutation alone conferred resistance only to NFV (Table) and, as expected, did not confer cross-resistance to other PIs. However, both phenotyping assays showed that D30N increased susceptibility to SQV (Fig. 1). The presence of N88D with D30N counteracted the HS to SQV and conferred a lower level of resistance (1.4-1.7X). Interestingly, viral isolates with both D30N and M46I remained hypersusceptible to SQV and there were no significant differences between this group and isolates with only D30N (p = 0.422). In addition, D30N also increased susceptibility to APV and the addition of N88D counteracted this effect. D30N also influenced the resistance to IDV caused by M46I; subtype B viral isolates with M46I had a mean FC to IDV of 4.7-6.1X compared to wild-type isolates (Fig. 1). Isolates with both D30N and M46I had a higher FC compared to wild-type isolates, but the resistance was 3.6-4.4X lower than isolates with M46I alone. We next evaluated the acquisition of DRMs across time of NFV exposure. In the first year of NFV-containing HAART, the major DRM was D30N alone (19%) and isolates with D30N/N88D, M46I, N88S or L90M were present in 4-7% of isolates (Fig. 2). By the third year of NFV exposure, D30N/N88D was the main DRMs corresponding to 31% of cases, while L90M and M46I were present in 21% and 13% of cases, respectively. V82A was the least frequent major DRM (2%), while D30N or N88S alone was observed in 8% and 6% of the samples, respectively. To better understand the persistence of D30N in the PR of patients under SQV/r and IDV treatment, we selected patients with previous NFV exposure containing only the D30N or D30N/N88D mutations. Follow-up of patients with previous NFV and subsequent APV usage was not feasible due to the low number of such cases. We hypothesized that if D30N counteracted resistance to SQV and IDV, selective pressures imposed by the latter drugs would favour D30N reversion or the development of a compensatory mutation. Indeed, of 16 initial patients with D30N alone that started an SQV/r containing-treatment, eight reversed codon 30 to asparagine and seven others acquired the compensatory DRM N88D (data not shown). Interestingly, of 11 isolates with an initial genotype of D30N/N88D, eight acquired L90M while maintaining D30N, and only three reversed to 30D after treatment with SQV/r. Treatment with IDV caused all seven isolates carrying only D30N to reverse this mutation. No isolates with an initial genotype of D30N/N88D with IDV-containing treatment were available for comparison. DISCUSSION In this study, we showed that the D30N mutation increases susceptibility to two PIs, SQV and APV and counteracts the resistance conferred by M46I to IDV. Other reports with phenotype data for D30N corroborated this HS (Patick et al. 1998, Rhee et al. 2004, 2006, Aoki et al. 2009), although that issue was not the focus of those studies. Here, we showed for the first time a significant disparity between the presence and absence of D30N alone or in combination with other DRMs in regards to the HS to other PIs. In HIV-1B isolates, D30N was the most frequent mutation selected by NFV-containing first-line HAART, as previously shown by other studies (Patick et al. 1998, Røge et al. 2003, Garriga et al. 2007). We showed that the preferential acquisition of D30N in subtype B isolates under NFV exposure is particularly prominent during the first year of treatment (Fig. 2). Unfortunately, the newer PIs darunavir and tipranavir could not be evaluated here because of the scarcity of phenotypic information available in public databases. Viral isolates containing D30N were also rarely tested for ATV because this drug was not available when most of the phenotyping assays were performed. The concept of increased susceptibility and drug re-sensitisation to improve outcomes of HIV viral load suppression in treated HIV/AIDS patients has already been clinically implemented with the combination of AZT and 3TC. The mutation M184V is known to confer resistance to 3TC and antagonistically prevent the acquisition of thymidine analogue mutations selected by AZT or d4T (Mouroux et al. 2001, Ait-Khaled et al. 2002). An alternative phenotype of a HS mutation is that it can suppress resistance caused by a DRM. The presence of the N88S mutation in the viral protease neutralizes resistance conferred by I50V to APV (Lam & Parkin 2003). In this study, the D30N mutation decreased the resistance caused by M46I to IDV and APV, although few isolates contained both D30N/M46I. However, Aoki et al. (2009) demonstrated the same phenotypic effect of D30N on viral susceptibility to APV when present alone and in combination with M46I. Such an effect is rarely seen with SQV because the mutations D30N and L90M are usually exclusive and require N88D to coexist in the same viral strain (Mitsuya et al. 2006). Here, we showed that N88D counteracted the HS caused by D30N to APV and SQV. Viruses with D30N have reversed this mutation in all patients following treatment with IDV. This also occurred in half of the patients treated with SQV/r, while other isolates harbouring D30N alone acquired the mutation N88D. We can envision a scenario in which the re-sensitisation effects of D30N could extend the time to DRM acquisition for IDV, SQV and APV, as shown for M184V and thymidine analogue mutations. We showed that the D30N mutation can cause increased susceptibility to APV and SQV while suppressing resistance conferred by M46I to IDV. Our data can be useful to better direct salvage therapy schemes with PI-containing HAART following NFV use, mainly in restrained resource countries where subtype B predominates, such as Latin America, northern Africa and China (Hemelaar et al. 2006). Patients with viruses containing D30N alone could be advised to use other boosted PIs such as SQV, IDV or APV because that mutation is specific for NFV resistance and causes HS to some PIs. A phase II clinical trial with a combination of NFV and APV has shown good safety and tolerability in HIV-1-infected patients (Eron et al. 2001) and could be a cost-effective alternative for these countries. However, additional studies are needed to evaluate the benefit of this combination in naïve-treatment patients and those under virologic failure. ACKNOWLEDGEMENTS This paper is part of the requirements for AFS to obtain his PhD degree at the Graduate Program in Genetics of the Universidade Federal do Rio de Janeiro, Brazil. REFERENCES
Copyright © 2011 - Instituto Oswaldo Cruz - Fiocruz The following images related to this document are available:Photo images[oc11029f2.jpg] [oc11029f1.jpg] [oc11029t1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}