 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Memórias do Instituto Oswaldo Cruz, Vol. 106, No. 2, 2011, pp. 194-199 ARTICLES Colorimetric microwell plate reverse-hybridization assay for Mycobacterium tuberculosis detection Candice Tosi MichelonI; Franciele RossoII; Karen Barros SchmidI; Rosa Dea SperhackeIII; Martha Maria OliveiraIV; Afrânio Lineu KritskiIV; Leonides Rezende JrV; Elis Regina Dalla CostaI, +; Andrezza Woloski RibeiroI; Mirela VerzaI; Patrícia Izquierdo CafruneII; Márcia Susana Nunes SilvaI, VI; Daniele KuhleisII; Arnaldo ZahaII; Maria Lucia Rosa RossettiI, VI ICentro
de Desenvolvimento Científico e Tecnológico, Fundação
Estadual de Produção e Pesquisa em Saúde, Porto Alegre,
RS, Brasil + Corresponding author: erdallacosta@yahoo.com.br Received 20 August
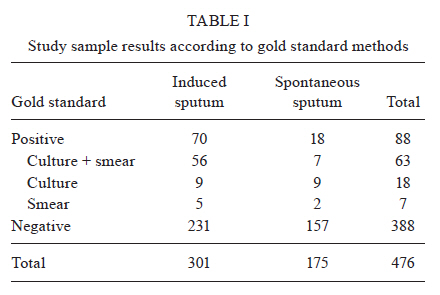
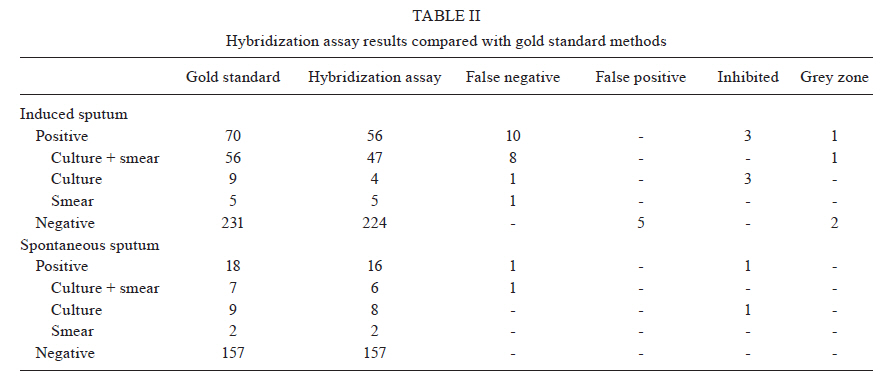
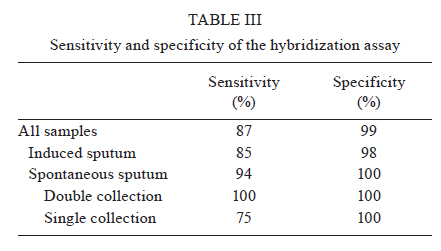
2010 Code Number: oc11032 ABSTRACT Direct smear examination using Ziehl-Neelsen staining for pulmonary tuberculosis (PTB) diagnosis is inexpensive and easy to use, but has the major limitation of low sensitivity. Rapid molecular methods are becoming more widely available in centralized laboratories, but they depend on timely reporting of results and strict quality assurance obtainable only from costly commercial kits available in high burden nations. This study describes a pre-commercial colorimetric method, Detect-TB, for detecting Mycobacterium tuberculosis DNA in which an oligonucleotide probe is fixed onto wells of microwell plates and hybridized with biotinylated polymerase chain reaction amplification products derived from clinical samples. The probe is capable of hybridising with the IS6110 insertion element and was used to specifically recognise the M. tuberculosis complex. When combined with an improved silica-based DNA extraction method, the sensitivity of the test was 50 colony-forming units of the M. tuberculosis reference strain H37Rv. The results that were in agreement with reference detection methods were observed in 95.2% (453/476) of samples included in the analysis. Sensitivity and specificity for 301 induced sputum samples and 175 spontaneous sputum samples were 85% and 98%, and 94% and 100%, respectively. This colorimetric method showed similar specificity to that described for commercially available kits and may provide an important contribution for PTB diagnosis. Key words: tuberculosis - reverse-hybridization assay - IS6110 - molecular diagnosis - oligonucleotide probe The World Health Organization reported 9.27 million new tuberculosis (TB) cases and 1.7 million TB-related deaths in 2007, 456,000 of which were among human immunodeficiency virus (HIV)-positive individuals. Undiagnosed pulmonary TB (PTB) infects, on average, 10-15 people every year (WHO 2009). Conventional tests for the laboratory confirmation of TB include acid-fast bacilli (AFB) smear microscopy, which can produce results in 24 h and bacterial culture (CDC 2009). Approximately 40-50% of patients with PTB are smear-positive (sputum must contain at least 5,000 bacilli/mL to be detectable by microscopy). Sputum smear microscopy is the primary tool for the diagnosis of TB in most countries. Although AFB microscopy is inexpensive, it lacks accuracy (sensitivity of 45-80% with culture-confirmed PTB cases), is labour-intensive and does not detect patients with smear-negative disease, mainly HIV-infected persons (Siddiqi et al. 2003, CDC 2009, WHO 2009). Bacterial culture is the most sensitive of the currently available tests and permits species identification and drug sensitivity tests. However, bacterial culture may require 2-8 weeks for the isolation of Mycobacterium tuberculosis from a clinical specimen. In addition, culture may give false-negative results in 10-20% of cases (Dye et al. 1999, Pai et al. 2004). The traditional gold standard for diagnosis is the combination of rapid identification of bacilli by direct microscopy with culture for subtype identification. However, given the diagnostic limitations of microscopy and the length of time required for traditional culture methods, attention has been focused on the development and evaluation of rapid methods for the detection of M. tuberculosis (Dinnes et al. 2007). To improve the case-detection rate, many new molecular techniques have been developed for TB diagnosis, including commercially available kits and in-house methods. Among them, nucleic acid amplification (NAA) has been shown to be a very rapid and sensitive method (Greco et al. 2006, Ling et al. 2008). Early laboratory confirmation of TB leads to earlier treatment initiation, improved patient outcomes, more opportunities to interrupt transmission and more effective public health interventions (CDC 2009). Nevertheless, many problems have been described during the use of in-house methodologies, including low reproducibility and subjective readings of the results (Kox et al. 1996). Our group has developed and evaluated an in-house polymerase chain reaction (PCR) using IS6110 as a target and two different procedures to detect M. tuberculosis complex DNA: agarose gel electrophoresis and membrane hybridization (Rossetti et al. 1997, Sperhacke et al. 2004, Verza et al. 2009). Because the sensitivity of PCR is dependent on the efficient preparation of clinical specimens before amplification and the detection method, this study describes a pre-commercial molecular test, Detect-TB (Ampligenix Biotech, Brazil), that includes an improved purification method for M. tuberculosis complex DNA and the development of a colorimetric method for its detection, in which an oligonucleotide probe is fixed into the wells of microwell plates and hybridized with biotinylated PCR amplification products derived from clinical samples. MATERIALS AND METHODS Bacterial strains and clinical specimens - The M. tuberculosis reference strain H37Rv (ATCC 27294) was used in the standardization of the extraction and purification procedure. A set of 120 pulmonary and extrapulmonary clinical samples from non-TB cases was used to determine the cut-off point of the reverse-hybridization assay. A total of 476 specimens from patients suspected of having TB were included to estimate the accuracy of the test. Among them, 301 had induced sputum specimens evaluated of which only one sample from each patient was processed. The remaining 175 specimens were spontaneous sputum samples, including 47 patients with a single collection that were processed in duplicate and 128 patients with two collections on different days that were processed at a single time. All samples (500 μL) were treated with 2% N-acetyl-L-cysteine (500 μL)/1 M NaOH (500 μL). Mycobacterial culture and smear microscopy were carried out for all clinical samples and the association of these results was chosen as the gold standard. Laboratory technicians who carried out molecular testing were blinded to the bacteriology results. This study was approved by the Ethical Committee of State Foundation for Production and Research in Health. DNA extraction and purification - DNA extraction and purification was standardized based on a protocol previously described by Boom et al. (1990). Some modifications were made to increase the sensitivity, reduce the extraction time and adapt the method to M. tuberculosis. A dilution containing approximately 50 colony-forming unit of the reference strain was used as the extraction positive control (EPC). A vessel containing 500 μL of a 1X TE buffer (10 mM Tris-HCl and 1 mM EDTA) was used as the extraction negative control (ENC). An EPC and an ENC were included with each set of samples (Sperhacke et al. 2004). Initially, a silica suspension was prepared. Six grams of silicon dioxide was suspended in demineralised water in a total volume of 50 mL in a glass cylinder and sedimented using gravity for 24 h at room temperature (RT). Forty-three millilitres of the supernatant was disposed by suction. Demineralised water was added to a total volume of 50 mL and the silica pellet was suspended by vigorous shaking. After 5 h of sedimentation at RT, 44 mL of the supernatant was disposed by suction and 60 μL of HCl (32% wt/vol) was added to adjust the suspension to pH 2.0. One millilitre aliquots were autoclaved for 20 min at 121ºC to destroy any contaminating nucleic acid and stored protected from light at 5ºC +/-3 (Gefrides et al. 2010). Samples were concentrated by centrifugation at 13,000 rpm for 10 min and the supernatants were disposed by suction. One hundred microlitres of lysis buffer (8 M guanidine hydrochloride, 0.08 M Tris-HCl, 0.04 M EDTA and 2% Triton X-100) were added and the samples were vortexed to homogeneity. After 10 min of incubation at 100ºC, the samples were centrifuged for 1 min at 13,000 rpm and the supernatants were transferred to a vessel containing 2.5 μL of the silica suspension previously prepared and then vortexed to homogeneity. After 1 min centrifugation at 13,000 rpm, supernatant was disposed by suction and the pellet was washed twice with a washing buffer (8 M guanidine hydrochloride and 0.08 M Tris-HCl) and once with 70% ethanol. After disposal of the ethanol, the vessels were dried out in a heat block at 56ºC for 10 min with closed lids and then at RT for 5 min with open lids. Thirty-three microlitres of 1X TE elution buffer was added and the vessels were vortexed briefly and incubated for 10 min at 56ºC. After 1 min of centrifugation at 13,000 rpm, 30 μL of the supernatant was collected and stored at -20ºC for further experimentation. DNA amplification - Amplification reactions were performed with biotinylated primers INS-1 (5'-CGTGAGGGCATCGAGGTGGC) and INS-2 (5'-GCGTAGGCGTCGGTGACAAA) derived from the IS6110 insertion element sequence, described by Hermans et al. (1990). PCR was performed in a final volume of 50 μL, containing 0.2 mM each of dATP, dCTP, dGTP and dTTP, 10 mM Tris-HCl (pH 8.0), 50 mM KCl, 3 mM MgCl2, 40 pmoles of each primer (INS-1 and INS-2) and 2.5 units of Taq DNA Polymerase (Invitrogen, Life Technologies, INC, Carlsbad, CA, USA). The amplification was carried out (35 cycles of 2 min at 94ºC, 2 min at 68ºC and 2 min at 72ºC) in an automated thermal cycler (PTC 150 Mini Cycler, MJ Research, Watertown, MA, USA). Amplified products were stored in the refrigerator for further experimentation. A negative control (PCR mixture with water instead of template DNA) and a positive control were included for each PCR set. The positive amplification control was a pAMP-1 plasmid containing a 245 bp inserted fragment from the IS6110 sequence (Cortez-Herrera et al. 2008). The specificity of the amplification was defined in a previous study (Hermans et al. 1990). To evaluate the presence of inhibitors in negative samples, an internal control (IC) (pAMP-1 plasmid containing a 664 bp inserted fragment) containing the same primer recognition sites as the INS-1 and INS-2 primers was added to the PCR (Cortez-Herrera et al. 2008). The sequence added was amplified using the same conditions as the 245-bp fragment, but it generated a 664-bp fragment. These results were detected by electrophoresis on a 1.5% agarose gel stained with ethidium bromide and visualized using UV light. Samples displaying inhibition were excluded from the analysis. Microwell hybridization assay - Amplicons were detected on microwell plates (Nunc Immobilizer™ Amino Surface, Nunc A/S, Roskilde, Denmark). The aminated probe (5'amine TTTTTTTTTTGCCCGTCCCGCCGATCTC3') was designed using Primer Express Software v2.0, (Applied Biosystems, CA, USA) and was complementary to an internal region of the amplified fragment. Microwell plates were prepared for detection according to manufacturer's instructions. One microgram of the probe was added to 100 μL of an immobilization buffer (100 mM Na2CO3, pH 9.6) and dispensed into microplate wells. Microwell plates were sealed with an adhesive cover (HotSeal, Diversified Biotech, Boston, USA) and incubated overnight at 2-8ºC. The probe was then removed and the wells were washed three times with phosphate buffered saline buffer (137 mM NaCl, 2.7 mM KCl, 3 mM Na2HPO4 and 10 mM KH2PO4, pH 7.4) containing 0.05% Tween-20 (Nunc Immobilizer™ Amino Instruction Protocol, 2005). One hundred microliters of hybridization buffer [5x saline sodium citrate (SSC), 0.5% bovine serum albumin (BSA), 0.1% Tween 20] and 30 μL of PCR products, previously denatured at 100ºC for 5 min and chilled on ice, were added to each well. Samples were reverse hybridized by incubating for 45 min at 50ºC. The wells were then washed three times at RT with a 5x SSC (2.9 M NaCl and 0.3 M C6H5Na3O7 :.2 H2O) containing 0.1% Tween-20 solution, soaked for 15 min with the same solution pre-heated to 50ºC and then washed three more times at RT. One hundred microliters of 0.1 M 4-morpholinepropanesulfonic acid buffer [400 ng/mL of streptavidin-peroxidase polymer, 30 mg/mL of BSA (fraction V), 20 mg/mL of mannitol, 60 mg/mL of saccharose, 1 mM CaCl2 and 0.2 mg/mL of thimerosal, pH 7.4] was added to each well and the plates were incubated for 30 min at 37ºC. The wells were washed three times with a Tris-buffered saline solution (100 mM Tris-HCl, 150 mM NaCl, 0.1% Tween-20), soaked for 5 min and then washed three more times with the same solution. One hundred microliters of 3,3',5,5'-tetramethylbenzidine (Moss, Pasadena, USA) was added. After incubation for 15 min at RT, 100 μL of 0.1 M H2SO4 was added to stop colour development. Absorbance was measured at 450 nm with a 620 nm re-filter using an enzyme-linked immunosorbent assay (ELISA) reader. Absorbance of the PCR negative control was subtracted from the results (Nunc 1999a, b). Cut-off determination - A pool of 120 pulmonary and extrapulmonary samples, TB-negative according to the gold standard, was used to determine the cut-off value of the reverse-hybridization assay. Samples with a grey zone reading were excluded from subsequent analysis. RESULTS The cut-off value of the assay was defined as the mean optical density plus three times the standard deviation value, resulting in a value of 0.250. The grey zone was defined as the values between 90% of the cut-off value and 10% above the cut-off value (0.225-0.275). Of the 301 induced sputum samples analyzed, 70 had positive results for culture or microscopy (5 only microscopy-positive, 9 only culture-positive and 56 positive for both), while 231 were negative for both. Of the 175 spontaneous sputum samples analyzed, 18 had positive results for culture or microscopy (2 only microscopy-positive, 9 only culture-positive and 7 positive for both), while 157 were negative for both (Table I). Of the 70 positive induced sputum samples, three showed inhibition in the PCR reaction, one had a reading in the grey zone and 56 showed positive results in the hybridization assay test; 10 had negative readings (false-negatives: 8 positive for microscopy and culture, 1 only microscopy-positive and 1 only culture-positive). From the 231 negative samples, two showed readings in the grey zone, 224 had negative results in the hybridization assay test and five had positive readings (false-positive) (Table II). After the analysis, the sensitivity and specificity were calculated to be 85% and 98%, respectively (Table III). Of the 18 positive spontaneous sputum samples, one showed inhibition in the PCR reaction, 16 showed positive results in the hybridization assay test and one (positive for microscopy and culture) had negative readings (false-negative). All of the 157 negative samples had a negative result in the hybridization assay test (Table II). The inhibited samples and the samples with readings in the grey zone were excluded from the analysis. After the analysis, the sensitivity and specificity were calculated to be 94% and 100%, respectively (Table III). Of the 175 spontaneous sputum samples, there were two groups of samples, one group of 128 that had two collections on two different days that were both processed at a single time and another group of 47 that had a single collection on only one day that was processed in duplicate. Of the 128 samples that had double collection, one (microscopy and culture-positive) showed inhibition in the PCR reaction for both specimens, 14 had positive results for culture or microscopy (2 only microscopy-positive, 7 only culture-positive and 4 positive for both) and 13 showed positive results in the hybridization assay test for at least one specimen, while 114 were negative for both tests. Of the group of 47 samples which had a single collection and were processed in duplicate, four had positive results for culture or microscopy (1 was only culture-positive and 3 were positive for both), of which three showed positive results least for one aliquot in the hybridization assay test and one (positive for microscopy and culture) had a negative reading (false-negative); the other 43 samples were negative for both tests. Sensitivity and specificity were calculated to be 94% and 100%, respectively, for spontaneous sputum. Sensitivity and specificity were calculated separately for the samples with double and single collection, being 100%, 100% and 75%, 100%, respectively (Table III). Of the 88 positive specimens, four showed evidence of inhibition. Their amplified products were detected using electrophoresis in a 1.5% agarose gel stained with ethidium bromide and visualized under UV light as two separated fragments, one of 664 bp, corresponding to the IC and another of 245 bp of the IS fragment. The inhibited specimens were tested as expected to not have the amplification of any fragment. These results represented an inhibition rate of 4.54% and were only observed among positive samples. DISCUSSION The hybridization assay was standardized for detection of M. tuberculosis-complex DNA derived from clinical samples. For this purpose, amplification products with the expected size of 245 bp (IS6110) were reverse-hybridized into wells of a microwell plate. This methodology was selected due to its simple method and the colorimetric end point, factors that might be expected to facilitate the transfer of NAA tests to laboratories in low-income countries (Dowdy et al. 2003, van Cleeff et al. 2005). This study describes a pre-commercial molecular test suitable for use as an alternative to currently available commercial kits. DNA extraction was based on the method described by Boom et al. (1990), with modifications to increase sensitivity. The overall sensitivity (87%) and specificity (99%) results obtained in this study are in agreement with the findings of other studies and are also comparable with commercial kits (Pai et al. 2003, Piersimoni & Scarparo 2003, Ling et al. 2008). The sensitivity and specificity using the spontaneous sputum samples was high; 94% and 100%, respectively. Among patients with no expectorated sputum, the sensitivity and specificity were 85% and 98%. Diagnosis of TB in these patients is difficult and, in most cases, these patients are treated empirically based on clinical and chest X-ray findings. Induced sputum evaluation has been proposed for those that cannot produce sputum spontaneously, but it has low sensitivity in sputum microscopy (Conde et al. 2000). It was not possible to repeat the analysis process using the 301 induced sputum samples due to the low volumes of sputum collected. Of these samples, three showed inhibition in the PCR reaction due to the presence of inhibitory substances that prevent or reduce amplification, causing false-negative results (Piersimoni & Scarparo 2003, Greco et al. 2006). These three samples were excluded from the analyses due to the fact that if inhibitors are present, the NAA test is of no diagnostic help for this specimen; we were unable to obtain additional samples from these patients. Of the 70 positive induced sputum samples, 10 were false-negative, probably due to uneven AFB distribution and their paucibacillary nature. Because of the inherent ability of mycobacteria to form clumps, clumping could have occurred in the aliquots processed for microscopy, culture and PCR and may have resulted in an insufficient number of detectable bacilli, which were suitable only for detection by smear or culture. In this case, an inappropriate specimen dilution during the decontamination procedure or a sampling error would reduce the test sensitivity, generating a false-negative result (Woods 2001, Piersimoni & Scarparo 2003, Chakravorty et al. 2005). Of the 231 negative induced sputum samples, five were false-positive, potentially due to cross-contamination that could have occurred during specimen processing, although we followed the guidelines for diagnostic methods, including unidirectional work flow comprising the physical separation of specimen processing and reagent preparation and the use of positive and negative controls during PCR amplification (Ersöz et al. 1998, Fukunaga et al. 2002, Chakravorty et al. 2005). Thus, the false positive results may also be due to the presence of non-cultivable bacteria in the specimen or to the high sensitivity of the PCR assay, which can even identify asymptomatic individuals infected with tuberculous bacilli (de Lassence et al. 1992, Chakravorty et al. 2005). Another important aspect of these false-positive results may come from the widely-used definition of positivity based on microbiological rather than clinical evidence (Scott et al. 2002, Piersimoni & Scarparo 2003). In the spontaneous sputum samples, one sample showed evidence of inhibitory activity and was excluded from the analysis and no false-positives were observed, providing a specificity of 100%. One of these samples was a false-negative, probably due to similar reasons as with the induced sputum, except for the potential pau-cibacillary nature. In samples collected using this method, we had two groups of patients: one with a single collection and samples processed in duplicate and another with two collections on different days and both samples processed at a single time. A significant improvement in the sensitivity was observed when double collection was performed. The data from the literature show a strong correlation between test sensitivity (within all categories of respiratory specimens) and the number of specimens tested for each patient, thus supporting this hypothesis that test performance is critically affected by the Mycobacteria burden and distribution within the sample (Piersimoni & Scarparo 2003). The use of in-house PCR methods for routine diagnosis has increased during recent years. However, impurities in DNA preparations may inhibit the reaction and even inert components can cause inhibition (Wilson 1997). In the diagnostic testing of highly prevalent infectious diseases, inhibitors responsible for false-negative results represent a serious problem. The specimens that contained PCR inhibitors, characterized by the absence of the targeted diagnostic fragments in positive samples, reinforced the need to overcome this problem by using an IC to effectively monitor the reaction (Almeda et al. 2000, Honoré-Bouakline et al. 2003). The use of the same primer sequences to amplify the M. tuberculosis diagnostic fragment and the IC fragment allowed the IC to be used without increasing the laboratory workload (Soini et al. 1992, Almeda et al. 2000). The NAA tests provided a reliable way of increasing the specificity of diagnosis to rule in disease. NAA tests could have the greatest impact in smear-negative disease, as the lower sensitivity makes ruling out disease more difficult. For smear-negative disease, the appropriate use of NAA tests will depend on the associated degree of clinical suspicion (Dinnes et al. 2007, CDC 2009). A positive NAA test is less likely to be a false-positive and, taken along with the full clinical picture, could help justify treatment; the NAA test result is one piece of evidence and would not determine the decision alone. There is a high degree of variability in accuracy across studies using NAA tests; thus, they cannot replace culture and microscopy but should be interpreted along with conventional tests and clinical data for diagnosing TB (Pai et al. 2003, 2004, Piersimoni & Scarparo 2003, CDC 2009). The lack of rapid and accurate diagnostic tests for TB has been a major concern for global TB control. The most notable advantage of NAA tests is their rapid turn-around time, which may have important implications for patient management and TB control (Dinnes et al. 2007). Compared with culture-based methods, NAA tests can detect the presence of M. tuberculosis bacteria in a specimen weeks earlier for 80-90% of patients suspected to have PTB, whose TB will be ultimately confirmed by culture (CDC 2009). Our NAA test may be less costly than currently available commercial tests and could be introduced more widely after a proper evaluation of its cost-effectiveness with clinical and radiographic characteristics to refine estimates of the likelihood of TB disease in different settings (Lim et al. 2000, van Cleeff et al. 2005). Our findings showed that the results of 95.2% of the tested samples in agreement to results using gold standard methods. The one discrepancy was potentially because the results of the study were influenced by the reference standard used. The lack of a diagnostic gold standard remains one of the major obstacles for evaluating new diagnostics. In summary, the colorimetric microwell reverse-hybridization method described in this study offers several advantages over alternative techniques. In addition to the reduced cost compared with current commercial kits, the results of this assay are based on spectrophotometry and do not depend on human interpretation (Kox et al. 1996). Furthermore, this assay is similar to ELISA in certain aspects, facilitating its application in routine laboratories in regions with high TB and TB/HIV burdens. REFERENCES
Copyright © 2011 - Instituto Oswaldo Cruz - Fiocruz The following images related to this document are available:Photo images[oc11032t1.jpg] [oc11032t3.jpg] [oc11032t2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}