 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Memórias do Instituto Oswaldo Cruz, Vol. 106, No. 6, Sept., 2011, pp. 705-715 Original Article Molecular phylogeny of the Myzorhynchella Section of Anopheles (Nyssorhynchus) (Diptera: Culicidae): genetic support for recently described and resurrected species Brian Patrick BourkeI; Sandra Sayuri NagakiI; Eduardo Sterlino BergoII; Jáder da Cruz CardosoI, III; Maria Anice Mureb SallumI, + IDepartamento
de Epidemiologia, Faculdade de Saúde Pública, Universidade de
São Paulo, Avenida Dr. Arnaldo 715, 01246-904, São Paulo, SP,
Brasil Received 28 February
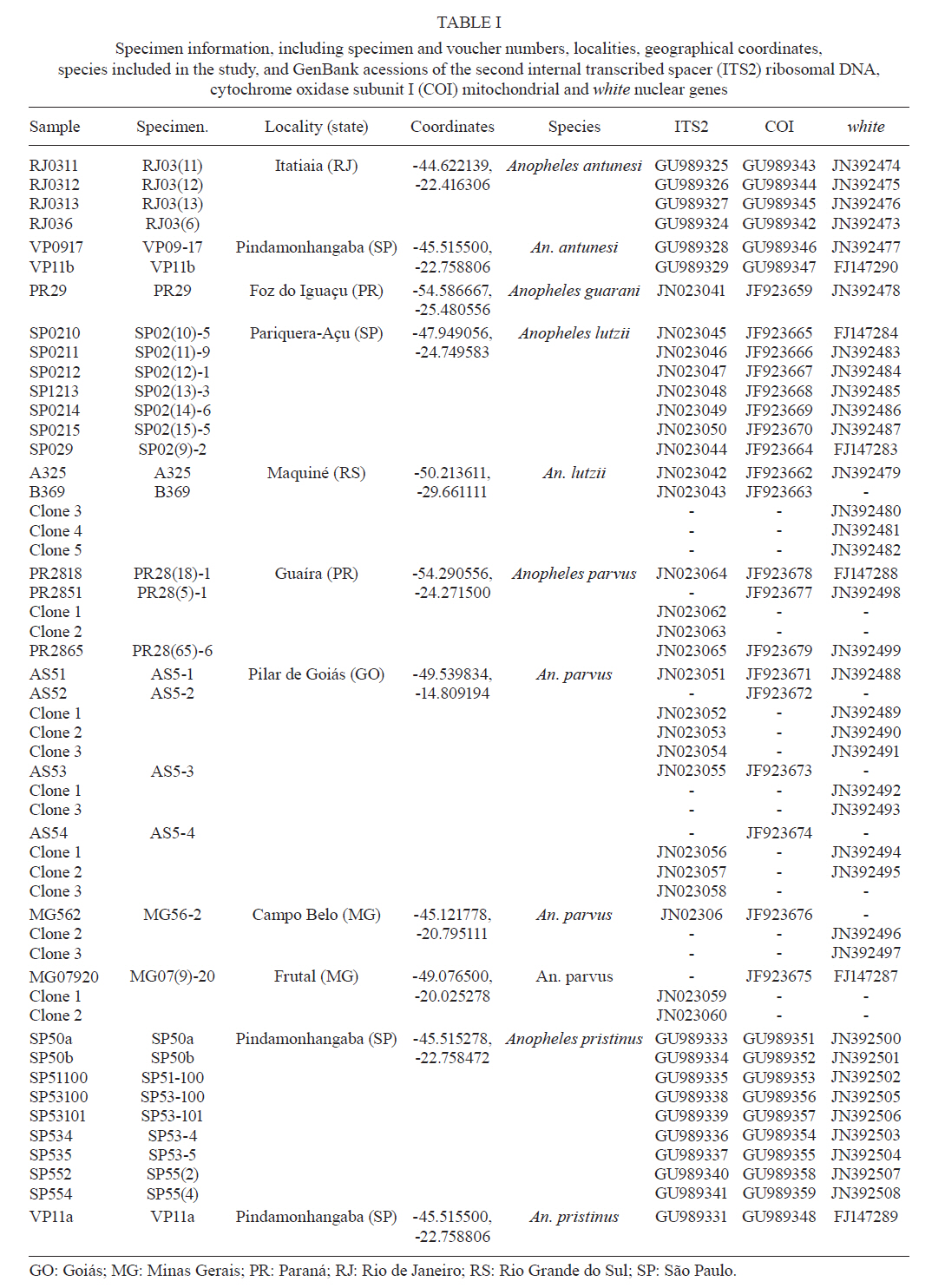
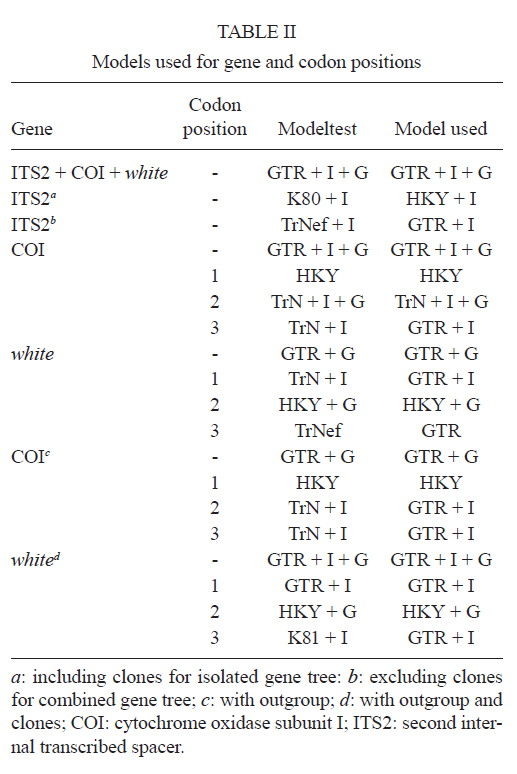
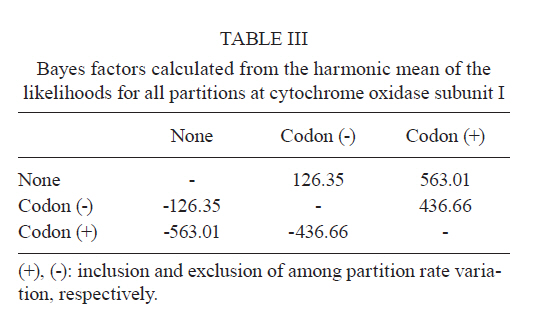
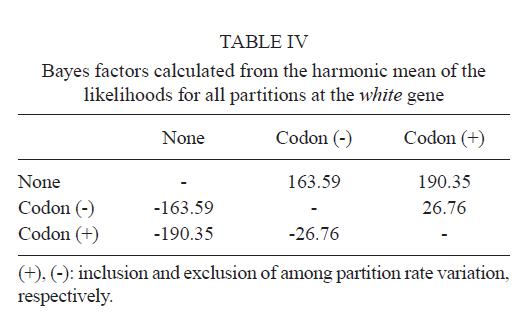
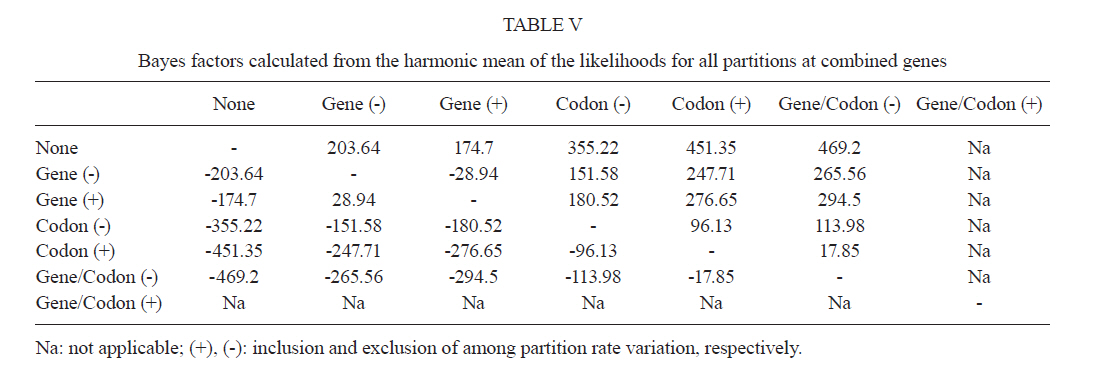
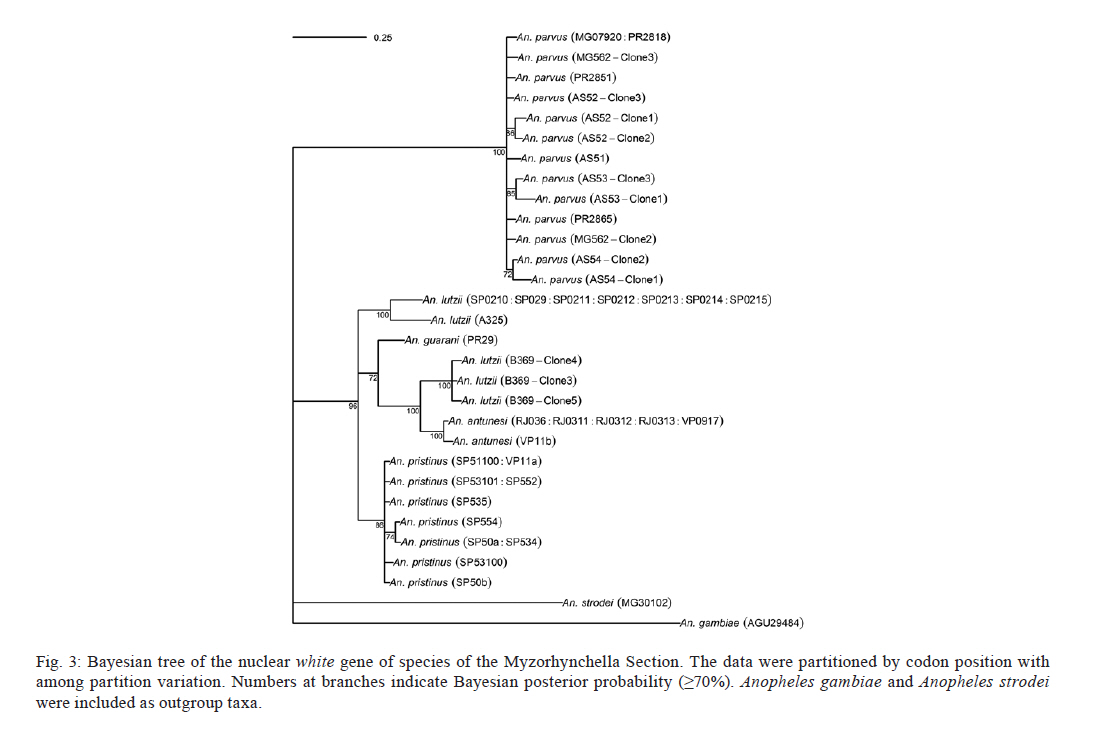
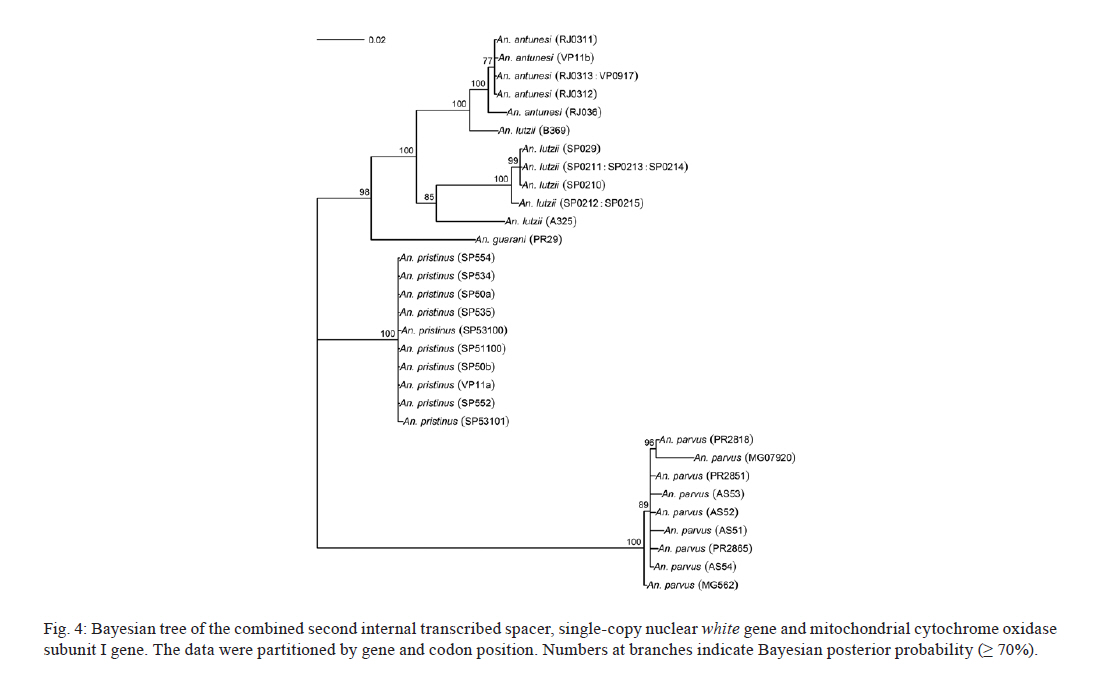
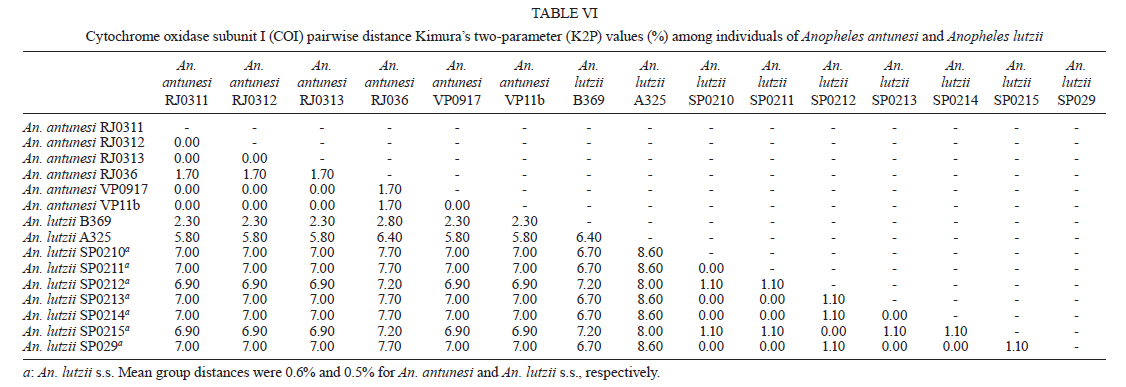
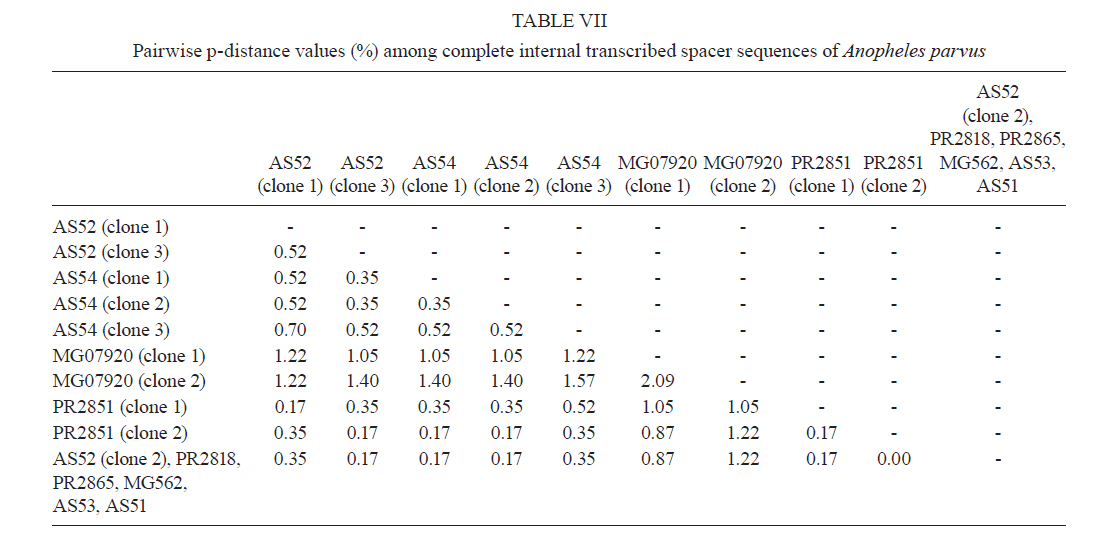
2011 Code Number: oc11120 Abstract Phylogenetic relationships among species of the Myzorhynchella Section of Anopheles (Nyssorhynchus) were investigated using the nuclear ribosomal DNA second internal transcribed spacer (ITS2), the nuclear whitegene and mitochondrial cytochrome oxidase subunit I (COI) regions. The recently described Anopheles pristinus and resurrected Anopheles guarani were also included in the study. Bayesian phylogenetic analyses found Anopheles parvus to be the most distantly related species within the Section, a finding that is consistent with morphology. An. pristinus and An. guarani were clearly resolved from Anopheles antunesi and Anopheles lutzii, respectively. An. lutzii collected in the same mountain range as the type locality were found within a strongly supported clade, whereas individuals from the southern state of Rio Grande do Sul, tentatively identified as An. lutzii based on adult female external morphology, were distinct from An. lutzii, An. antunesi and from each other, and may therefore represent two new sympatric species. A more detailed examination of An. lutzii sensu latoalong its known geographic range is recommended to resolve these anomalous relationships. Key words: phylogeny, Anopheles, Myzorhynchella, white, ITS2, COI Mosquitoes (Culicidae) are highly diverse geographically widespread taxa containing approximately 3,529 species (Harbach 2011a). They are also of major medical importance as a consequence of being vectors of pathogens that cause diseases in humans such as dengue, filariasis and malaria (WHO 1989). All known vectors of malarial parasites belong to the genus Anopheles Meigen and are responsible for an estimated 243 million malaria cases annually (WHO 2009). Within the Americas there are an estimated one million cases of malaria annually, the majority of which are caused by vectors of the subgenus Nyssorhynchus Blanchard (Zimmerman 1992, WHO 2009). This subgenus contains 39 species (Harbach 2011b, Nagaki et al. 2011), at least seven of which are known to harbour malarial parasites (Rosa-Freitas et al. 1998). Whereas the subgenus is a well supported monophyletic group (Sallum et al. 2000, 2002, Harbach & Kitching 2005), relationships among species within this group are less well defined. The subgenus has traditionally been divided into three sections based on morphological characters (Peyton et al. 1992): the Argyritarsis, Albimanus and Myzorhynchella Sections. Until recently the Myzorhynchella Section was comprised of four nominal species, Anopheles lutzii Cruz, Anopheles parvus (Chagas), Anopheles nigritarsis (Chagas) and Anopheles antunesi Galvão & Amaral (Harbach 2004). The Myzorhynchella Section is currently the only section in Nyssorhynchus without species implicated in malaria transmission and the only section with some genetic support as a natural grouping. Genetic data also indicate that species diversity within the Myzorhynchella Section is perhaps underestimated due to the existence of species complexes (Bourke et al. 2010). Nagaki et al. (2011) recently resurrected Anopheles guarani from An. lutzii and described a new species, Anopheles pristinus Nagaki & Sallum, distinguished from An. antunesi. Members of species complexes frequently vary in their ability to transmit malaria and an effective assessment of mosquito susceptibility to the malaria parasite relies on the ability to accurately resolve and identify species. Recent studies have worked towards identifying malaria refractory genes in Anopheles (Marshall & Taylor 2009, Corby-Harris et al. 2010). If the Myzorhynchella Section is a natural grouping within Nyssorhynchus without vectors of human malarial protozoa, it may prove to be a useful group for the study and identification of such genes. The current study therefore seeks to describe the phylogenetic relationships among species within the Myzorhynchella Section, assess the validity of recently described An. pristinus and An. guarani and determine the species status of anomalous individuals using the nuclear ribosomal DNA second internal transcribed spacer (ITS2), the mitochondrial cytochrome oxidase subunit I (COI) and the single copy nuclear white gene. Materials and Methods Mosquito collection - A description of the specimens used in this study can be found in Table I. These specimens included the offspring of females caught in the field using a Shannon trap (Shannon 1939) and larvae and pupae collected from immature habitats, which were then raised to adulthood. Species identification of all but two specimens was based on either adult male genitalia or fourth-instars larval characteristics. Specimens An. lutzii A325 and An. lutzii B369 were identified only by female morphology. DNA extraction - DNA was extracted from each specimen according to the animal tissue DNA extraction protocol provided by the QIAgen DNeasy® Blood and Tissue Kit (QIAGEN Ltd, Crawley, UK). All extractions were diluted to 200 µL with the buffer provided and extraction solutions were retained for storage at -80ºC in the entomological frozen collection of the School of Public Health, University of São Paulo, Brazil. ITS2 region - This region was amplified using the 5.8SF (5'-ATCACTCGGCTCGTGGATCG-3') and 28SR (5'-ATGCTTAAATTTAGGGGGTAGTC-3') primers using the same protocol adopted by Sallum et al. (2008). The polymerase chain reaction (PCR) was conducted in a total volume of 25 µL containing 1 µL of DNA extraction solution, 1 × PCR buffer (Invitrogen),1.5 mM MgCl2 (Invitrogen), 1.25 µL dimethly sulfoxide (Sigma), 0.1 µM of each primer, 200 mM each dNTPs (Amresco) and 1.25 U Taq Platinum polymerase (Invitrogen). The reaction proceeded under the following temperature regime: 94ºC for 2 min, 34 cycles of 94ºC for 30 s, 57ºC for 30 s and 72ºC for 30 s and a final extension at 72ºC for 10 min. COI mitochondrial gene - This region was amplified using LCO1490 (5'-GGTCAACAAATCATAAAGATATTGG-3') and HCO2198 (5'-TAAACTTCAGGGTGACCAAAAAATCA-3') primers (Folmer et al. 1994). DNA working solution was first made by diluting the DNA extraction solution to 1:20 using ultra-pure autoclaved water. The PCR reaction was conducted as above for ITS2. The reaction proceeded under the following temperature profile: 95ºC for 2 min, 35 cycles of 94ºC for 1 min, 57ºC for 1 min and 72ºC for 1 min and a final extension at 72ºC for 7 min. White nuclear gene - This gene was amplified using WZ2E and WZ11 primers (Besansky & Fahey 1997). This amplification product then served as a template in a sequencing reaction using internal primers W1F (5'-GATCAARAAGATCTGYGACTCGTT-3') and W2R (5'GCCATCGAGATGGAGGAGCTG-3'). The initial PCR reaction contained approximately 3 µL of DNA extraction solution in a total volume of 25 µL containing 1 × PCR buffer (Invitrogen), 1.5 mM of MgCl2 (Invitrogen), 200 mM of each dNTP (Amresco), 2 µM of each primer and 0.625 U Taq DNA Polymerase (Invitrogen). The reaction proceeded under the following temperature regime: 94ºC for 5 min, 35 cycles at 94ºC for 30 s, an annealing temperature of 50ºC for 1 min and then 72ºC for 2 min followed by a final extension at 72ºC for 10 min. The next step then involved taking this PCR product for sequencing using the W1F and W2R internal primers. Cloning - ITS2 PCR amplicons obtained from An. parvus were purified using PEG precipitation (20% polyethylene glycol 8000/2.5 M NaCl) and cloned into pGem-T Easy Vector (Promega, Madison, WI, USA). Three positive clones were sequenced. For the nuclear white gene, females were cloned when the direct sequence showed either unreadable peaks or double peaks. Individuals that had either ITS2 or white gene cloned are shown in Table I. Sequencing and sequence alignment - Sequencing reactions were carried out in both directions using a Big Dye Terminator cycle sequencing kit v3.1 (Applied Biosystems) and Applied Biosystems 3130 DNA Analyzer (Applied Biosystems). The COI and white gene sequences were aligned first by nucleotides using ClustalX (Thompson et al. 1997) and then by amino acid using TranslatorX (Abascal et al. 2010). The intron in the white gene could not be reliably aligned and so was excluded from further analyses. In addition, only a single randomly selected sequence was used to represent cloned individuals in the combined gene analysis. In the case of ITS2, sequences were first annotated for the 5.8S and 28S ends. Template ITS2 secondary structure was then predicted for An. antunesi RJ036 with Predict ITS2 Structure in the ITS2 Database (Koetschan et al. 2010). The remaining secondary structures were modelled at this source with Custom Modelling using the predicted An. antunesi RJ036 ITS2 secondary structure (above) as a template. Sequences with secondary structures were then aligned and edited in 4Sale (Seibel et al. 2006, 2008). It was not possible to align the complete ITS2 sequences and so those regions that could not be aligned were excluded from further analysis. Additionally, we were unable to align ITS2 sequences with an appropriate outgroup and so trees constructed using ITS2 sequence data (including the combined gene trees) are unrooted. Phylogenetic analyses - Bayesian analyses were applied to ITS2, COI and white sequence data using a partitioning strategy to allow different partitions to have their own model characteristics (composition, rate matrix and among-site variation) and to allow for among-partition rate variation. Datasets could be left unpartitioned, partitioned by gene, partitioned by codon position or partitioned by both gene and codon position. Optimal evolutionary models were determined for isolated partitions using the Akaike Information Criterion (AIC) in Modeltest (Posada & Crandall 1998). Optimal models for partitioned data were calculated using Bayes factors (BF) according to the formula BF21 = 2[ln(HM2) - ln(HM1)], where HM2 and HM1 are the harmonic means of the posterior sample of likelihoods from the pair of partition strategies being compared (Brown & Lemmon 2007). Positive values of BF21 are indicative of support for the partition denoted two over the partition denoted one. A BF21 value greater than 10 indicates significant support for partition two, values between 10 and -10 indicate ambiguity and values less than -10 indicate significant support for partition one. All Bayesian analyses were performed using MrBayes (Ronquist & Huelsenbeck 2003) and each analysis consisted of two runs to provide confirmation of convergence of posterior probability distribution. For COI, white, ITS2 and all but one of the combined analyses of gene partition strategies, each run was four million generations long and the first two million were discarded as burn-in. The Metropolis-coupled Markov chain Monte Carlo strategy was used with four heated chains; adequate mixing was achieved by setting the chain temperature to 0.1 for isolated genes and combined genes. Convergence of topology between the two runs was monitored using the average standard deviation of split frequencies - this index consistently fell to below 0.015 in the post-burn-in samples. Convergence was also monitored by noting that the potential scale reduction factor values were all approximately 1.0 in the post-burn-in samples. The one exceptional partition strategy (partitioning by gene and codon position with among partition rate variation) did not converge (runs were varied from two-40 million generations, with four-eight heated chains and chain temperatures from 0.05-0.25) and so was excluded from further analysis. Post burn-in samples were then used to construct a consensus tree containing nodes with greater that 70% posterior probability support. Trees were drawn using the R package APE (Paradis et al. 2004). Pairwise genetic distances for COI were calculated in MEGA4 (Tamura et al. 2007) using Kimura's (1980) two-parameter (K2P) model. Uncorrected pairwise p-distances were calculated in MEGA4 from a separate An. parvus ITS2 alignment using ClustalX. RESULTS The alignment included sequences from 35 individuals. Difficulties with aligning ITS2 (at positions 143-199, 223-285, 386-635 and 714-797) and the intron in the white gene resulted in these positions being excluded from the phylogenetic analyses. Non-overlapping sites from the white gene were also excluded. In total, 1,539 sites were included in the analyses, consisting of 363 sites from ITS2, 657 sites from COI and 519 sites from the white gene. GenBank accessions of the nuclear white, mitochondrial COI and the ITS2 are in Table I. Anopheles gambiae Giles and Anopheles strodei Root were used as outgroup taxa in the analyses of COI and white gene. It was not possible to find an appropriate outgroup for the ITS2 sequences and so ITS2 and combined COI-white-ITS gene trees were unrooted. The optimal models of evolution determined for each sequence partition are displayed in Table II. Where the selected rate matrix was unavailable in MrBayes (i.e. TrN and TIM), the most similar rate matrix available was selected (i.e. GTR). ITS2 is a non-coding region and so was left unpartitioned in the analysis. The best evolutionary model for this region was the TrNef+I model (substituted with the GTR+I model in Bayesian analyses). BF (Tables III, IV, V) found the optimal partitioning strategies for COI, white and combined genes. The best model for the COI gene (with an outgroup taxa) was one that partitioned the data by codon position and included among-partition rate variation (APRV) (Table III). The model chosen for the white gene (with an outgroup taxa) was one that partitioned the data by codon position and included APRV (Table IV). The model chosen for combined genes was one that partitioned the data by gene and codon position (Table V). The results of Bayesian analyses show a high degree of congruence among the ITS2, COI, white and combined gene trees (Figs 1, 2, 3 and 4, respectively). An. parvus, An. pristinus and An. guarani are strongly supported as species across all trees. In addition, the majority of the An. lutzii individuals (7 of 9) form a strongly supported clade across all trees with support of 100% Bayesian posterior probability (BPP). Some incongruence also exists between gene trees. The remaining two An. lutzii individuals (A325 and B369) cluster differently across trees. In the white and combined gene trees (Figs 2, 4), individual A325 forms a clade with the An. lutzii sensu stricto group described above (> 85% BPP), whereas the other (represented by the 3 clones from B369) forms a clade with An. antunesi (100% BPP). Both individuals form a clade with An. antunesi at the COI gene (78% BPP) (Fig. 2), whereas A325 was unclustered with An. lutzii at ITS2 (Fig. 1). The COI and white gene trees (Figs 2, 3) were rooted with an outgroup. In the white gene tree (Fig. 3), An. strodei and An. parvus were sisters to a clade containing the remaining species in the Myzorhynchella Section (96% BPP). In the COI tree (Fig. 2), An. strodei and An. parvus were sisters to An. pristinus, An. guarani and the An. lutzii-An. antunesi clade. In the combined gene tree (Fig. 4), An. pristinus was sister to a clade containing An. guarani, An. antunesi and An. lutzii (98% BPP) and within this clade An. guarani was sister to a clade containing all individuals of An. antunesi and An. lutzii (100% BPP). COI pairwise genetic distances among An. lutzii and An. antunesi were calculated under the K2P model (Table VI). Pairwise distances among individuals within An. antunesi and An. lutzii s.s. were found to be less than 2%, whereas differences between individuals from each group were in the range 6.90-7.70%. Individuals An. lutzii A325 and An. lutzii B369 were most distant to those from the An. lutzii s.s. group (8.00-8.60 and 6.40-7.20%, respectively). The distances between A325 and An. antunesi individuals ranged from 5.80-6.40, whereas the distances between B369 and An. antunesi ranged from 2.30-2.80%. An. lutzii A325 and An. lutzii B369 differed from each other by 6.4%. Difficulty in aligning ITS2 sequences resulted in large numbers of polymorphic sites being excluded from the phylogenetic analyses. It is likely the exclusion of such sites contributed to the existence of the large polytomy observed among An. parvus individuals and the lack of resolution for An. lutzii s.s. and An. lutzii A325. A separate alignment of An. parvus individuals was constructed to include all sites and pairwise p-distances were calculated to describe variation among these individuals (p-distances) (Table VII). An. parvus was the only species in the study to have intragenomic variation at ITS2. An. parvus showed higher intragenomic (0.17-2.09%) than intraspecific (0-1.57%) variation. Two clones from a single individual from the state of Minas Gerais (MG) differed from each other by 2.09% and from other conspecifics by 0.87-1.57%. This same individual was also clearly resolved from the remaining An. parvus individuals at the COI gene. A separate alignment of An. lutzii and An. antunesi could not be constructed because of the frequency of ambiguous sites and so p-distances could not be calculated as in the case of An. parvus. Instead, we calculated the proportion of unambiguously aligned sites for these ITS2 sequences on a pairwise basis using secondary structure (Table VIII). An. lutzii and An. antunesi consisted of four unique ITS2 sequences, one each for An. lutzii A325, An. lutzii B369, An. antunesi and An. lutzii s.s., giving a total of six pairwise combinations. The results indicated that the An. lutzii A325 and An. lutzii B369 sequence pair was the most difficult to align with only 77% of sites aligned, compared to 0.91% for An. lutzii s.s. and An. antunesi. Discussion There is scant molecular data available for species of the Myzorhynchella Section. However, in Bourke et al. (2010) phylogenetic analysis of the subgenus Nyssorhynchus, the Myzorhynchella Section was found to be monophyletic (at white and combined white-ND6 genes) and An. parvus was consistently found to be the sister to the remaining species of the Myzorhynchella Section. Our study found An. parvus to be sister to the Myzorhynchella clade and An. strodei in the white gene tree, whereas at COI it was found to be sister to An. pristinus, An. guarani, an An. antunesi-An. lutzii sensu lato clade and An. strodei from the outgroup. An. parvus was also the most distantly related species within the Section based on branch lengths. This distinct relationship is also supported by morphological characters that distinguish species within the Myzorhynchella Section. An. parvus can be separated from the remaining species in the Section by a distinct hook-like apex at the aedeagus in the male genitalia (Root 1927, Galvão 1941). Additionally, the eggs of An. parvus have an anterior crown-like structure and float vertically, characteristics which distinguish the species not only from species of the Myzorhynchella Section but also from Anopheles species in general (Forattini et al. 1998). Intraspecific variation did not support An. parvus as a species complex. However, a single individual from MG did show some evidence of distinction from the remaining individuals of An. parvus, as well as high intragenomic variation at ITS2. Concerted evolution normally maintains fixed interspecific differences and intraspecific homogeneity within ribosomal multigene families (Arnheim 1983). However, intragenomic variation occurs when the rate of mutation exceeds the rate of homogenization. Although fixed interspecific differences and intraspecific homogeneity at ITS2 has permitted unambiguous species identification in a range of closely related Anopheles species (Collins & Paskewitz 1996, Beebe et al. 2001, Wilkerson et al. 2004, Li & Wilkerson 2005), the intraspecific variation in ITS2 that sometimes occurs within Anopheles can pose a major problem for population and phylogenetic studies when it exceeds variation between populations. Such markers are likely to be of little use for the resolution of populations and may even lead to the misidentification of species (Li & Wilkerson 2007). Here we find higher sequence variation within an individual from MG than that found between all other individuals of An. parvus. In addition, both sequences in this individual were very different from all other sequences in An. parvus. Although this finding may be suggestive of population or taxonomic differences in An. parvus, given the extent of intragenomic variation, we consider the utility of ITS2 to be limited in more detailed future phylogenetic analysis of this species. There is general consistency in the remaining species relationships found at all genes, i.e. the existence of An. lutzii and An. antunesi species complexes and support for the recently described An. guarani and resurrected An. pristinus. An. lutzii was first described from individuals collected from the state of Rio de Janeiro (Cruz 1901). A later study synonymised An. niger Theobald and An. guarani with An. lutzii (Lane 1953). Consequently, the geographic distribution for this species became quite extensive, with records from Argentina, Brazil, Mexico and Paraguay. Recently, Nagaki et al. (2011) found morphological support for the resurrection of An. guarani from An. lutzii and for An. niger to be synonymised with An. guarani. Indications are that, contrary to having a continental-scale distribution, An. lutzii may be restricted to the Atlantic Forest of southeastern Brazil (Nagaki et al. 2011). Our analysis again found support for the distinction of An. guarani. An. lutzii on the other hand is found to be paraphyletic with respect to other species in the group. Whereas most An. lutzii individuals [from the state of São Paulo (SP)] consisted of one strongly supported group likely to be An. lutzii s.s. (with the type specimen originating from the same Serra do Mar mountain range), the lack of monophyly in this species was caused by two individuals tentatively identified as An. lutzii collected from the most southerly Brazilian state of Rio Grande do Sul. These two individuals were found to consist of two undescribed sympatric species, one of which appeared more closely related to An. antunesi and the other which was associated with both the An. lutzii s.s clade and An. antunesi. The COI genetic distances observed between An. lutzii A325 and An. antunesi (5.80-6.40%) and An. lutzii s.s (8-8.60%) individuals are approximately 10 times greater than mean within group distances (0.60% and 0.50% for An. antunesi and An. lutzii s.s, respectively). This finding is consistent with the sequence threshold of 10 times the mean intraspecific variation to delimit animal species proposed by Hebert et al. (2004). Although the relationship between An. lutzii B369 and An. antunesi falls outside this threshold (4 times mean intraspecific variation), genetic distances between An. lutzii B369 and An. antunesi individuals (2.30-2.80%) remained high and were close to the 3% threshold used by Hebert et al. (2003) to resolve 196 of 200 species of Lepidoptera. Fixed interspecific differences and intraspecific homogeneity generally found at ITS2 have proved effective at resolving many closely related Anopheles species and our results found that An. antunesi and An. lutzii s.s. are each represented by a single ITS2 sequence. Differences observed between An. lutzii A325 and An. lutzii B369 (based on the proportion of sites successfully aligned) were greater than interspecific differences. However, the exclusion of a large numbers of potentially important sites at ITS2 from phylogenetic analysis may have accounted for the poor resolution between An. lutzii A325 and An. lutzii in the ITS2 tree. It is notable that An. lutzii A325 and An. lutzii B369 were the only individuals in this study to be identified solely by adult female morphology. An. lutzii can be differentiated from other members of the Myzorhynchella Section by having small patches of pale scales at the proximal and distal ends of two white spots on the vein R4+5 of the wing (versus other combinations of dark and white spots) (Nagaki et al. 2011). However, important differences in the egg, larval and male genitalic morphology may exist that differentiate them from An. lutzii and An. antunesi. From its original description by Galvão and Amaral (1940) from SP, An. antunesi has been recorded across a large geographical range from northeastern Brazil (Rebêlo et al. 2007) to Argentina (Gorham et al. 1967, Darsie 1985) and Uruguay (Rodriguez & Varela 1962, Gorham et al. 1967). However, recent examination of An. antunesi from the type locality has shown that individuals formerly described as An. antunesi can be resolved into two sympatric species, An. antunesi and An. pristinus, based on the pattern of pale and dark wing spots, male genitalia and fourth-instar larva (Nagaki et al. 2010). Our analyses support this finding by resolving An. antunesi and An. pristinus and providing strong support for the monophyly of An. pristinus. However, we also found An. antunesi clusters with An. lutzii, as mentioned earlier, which is a relationship that has been recovered in a previous study of Nyssorhynchus phylogeny (Bourke et al. 2010). As a result of these findings, we find it necessary to question the status of An. antunesi in much of its reported range and suggest that the reports of the species in these varied localities may be the result of misidentifications. The main findings of the current study confirm the species status of An. pristinus and An. guarani and identify a strongly supported An. lutzii s.s clade and two species complexes (An. antunesi and An. lutzii complexes). To further clarify phylogenetic relationships among species within the Myzorhynchella Section, we propose additional sampling and morphological analyses (egg, larval, pupal, male genitalic and adult female morphology) of An. lutzii s.l. from various localities in southern and southeastern Brazil. These individuals may then be more accurately identified, as a particular form or species, prior to additional phylogenetic analysis. In addition, the findings of Nagaki et al. (2010, 2011) and the current study underlines the need for a reevaluation of the geographic distribution of the species of the Myzorhynchella Section in general. The principal questions raised from this study, therefore, are whether published records of An. antunesi from outside the type locality, such as Argentina and Uruguay, refer to the nominate species or to An. pristinus and, similarly, whether reports of An. lutzii to date refer to An. lutzii s.s., to a distinct species in an An. antunesi complex, or should be classified as An. guarani. The reports of An. lutzii from Mexico are a good example of potential confusion associated with this species. The individuals of An. niger, originally described as Myzorhynchella nigra by Theobald (1907) and synonymised with An. lutzii (Chagas 1907, Belkin 1968), are now potentially An. guarani, as noted by Nagaki et al. (2011). Consequently, resolving the apparent complexes and undertaking a morphological re-examination of individuals identified in collections as An. antunesi and An. lutzii will be the basis for providing more accurate distributions of species in the Myzorhynchella Section. Acknowledgements To the three anonymous reviewers, for their great contribution to the improvement of the text. References
Copyright © 2011 - Instituto Oswaldo Cruz - Fiocruz The following images related to this document are available:Photo images[oc11120t5.jpg] [oc11120f4.jpg] [oc11120t3.jpg] [oc11120t2.jpg] [oc11120t1.jpg] [oc11120t4.jpg] [oc11120t7.jpg] [oc11120f2.jpg] [oc11120t8.jpg] [oc11120f1.jpg] [oc11120f3.jpg] [oc11120t6.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}