 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
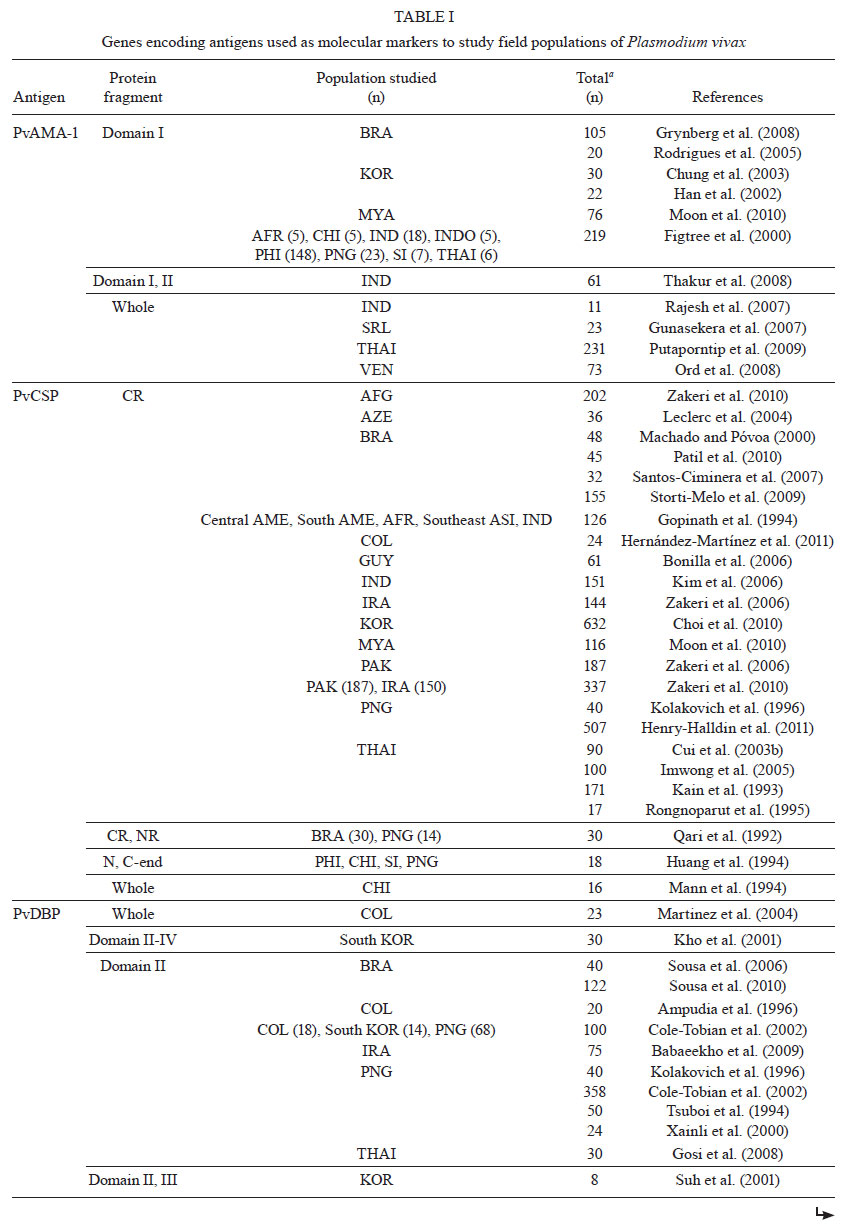
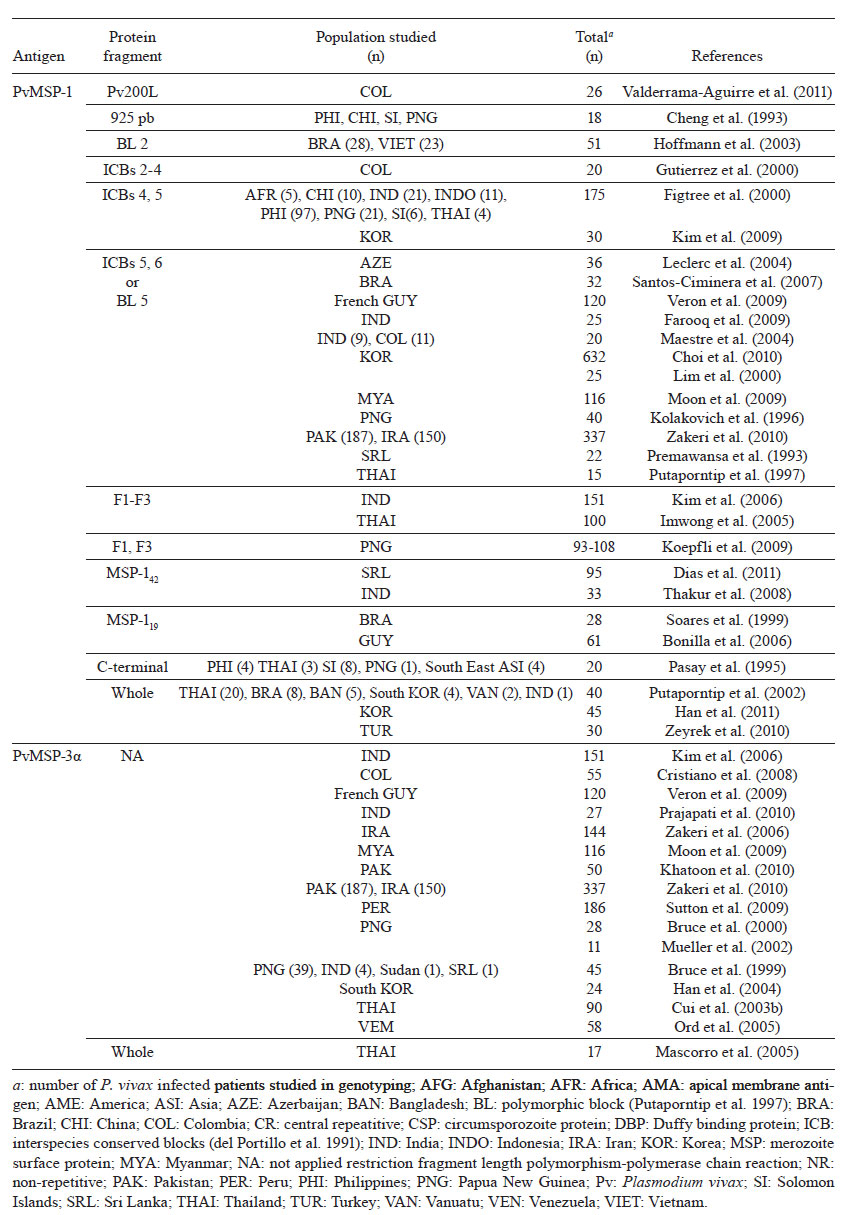
Memórias do Instituto Oswaldo Cruz, Vol. 106, Special Issue, pp. 12-26 Original Article Molecular markers and genetic diversity of Plasmodium vivax Cristiana Ferreira Alves de BritoI, +; Marcelo Urbano FerreiraII ILaboratório
de Malária, Instituto de Pesquisas René Rachou-Fiocruz, Av. Augusto
de Lima 1715, 30190-002 Belo Horizonte, MG, Brasil Received 26 April
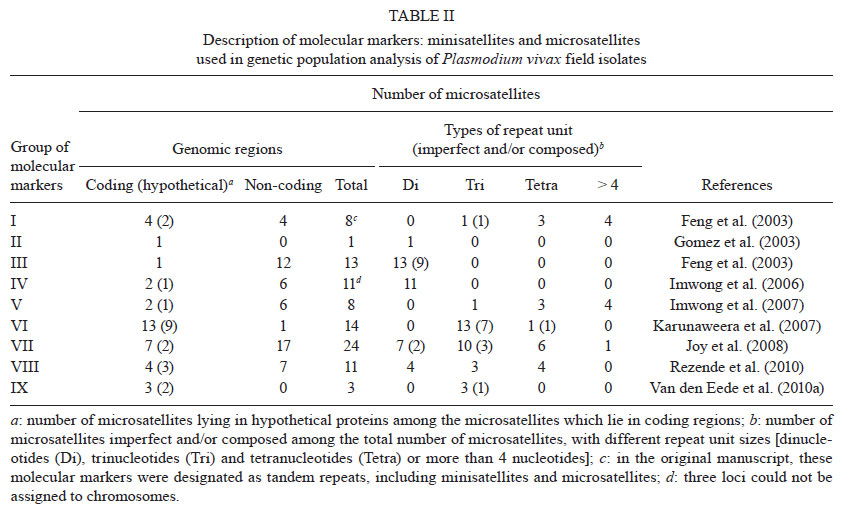
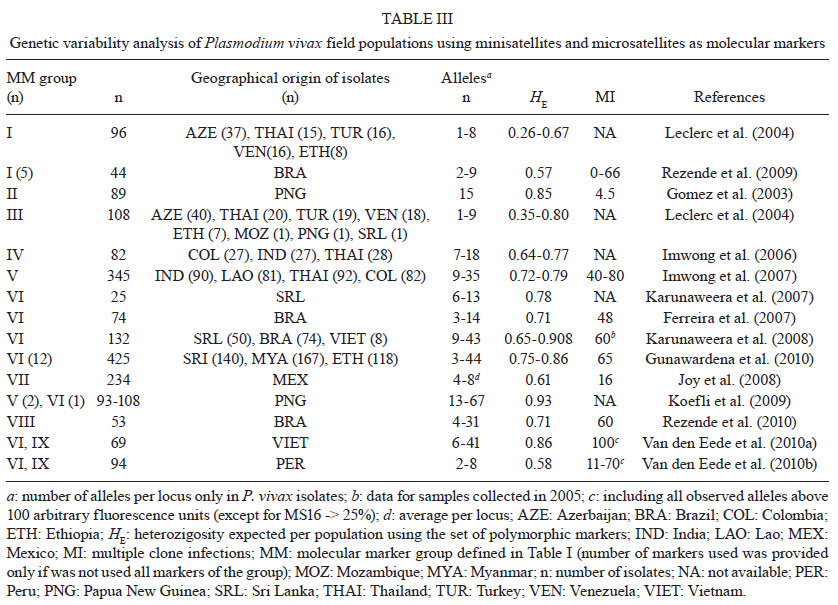
2011 Code Number: oc11136 Abstract Enhanced understanding of the transmission dynamics and population genetics for Plasmodium vivax is crucial in predicting the emergence and spread of novel parasite phenotypes with major public health implications, such as new relapsing patterns, drug resistance and increased virulence. Suitable molecular markers are required for these population genetic studies. Here, we focus on two groups of molecular markers that are commonly used to analyse natural populations of P. vivax. We use markers under selective pressure, for instance, antigen-coding polymorphic genes, and markers that are not under strong natural selection, such as most minisatellite and microsatellite loci. First, we review data obtained using genes encoding for P. vivax antigens: circumsporozoite protein, merozoite surface proteins 1 and 3α, apical membrane antigen 1 and Duffy binding antigen. We next address neutral or nearly neutral molecular markers, especially microsatellite loci, providing a complete list of markers that have already been used in P. vivax populations studies. We also analyse the microsatellite loci identified in the P. vivax genome project. Finally, we discuss some practical uses for P. vivax genotyping, for example, detecting multiple-clone infections and tracking the geographic origin of isolates. Key words: malaria - Plasmodium vivax - genetic markers - genetic diversity - multiple-clone infections - microsatellites Plasmodium vivax, a relatively neglected human malaria parasite, is a major public health challenge for Central and South America, the Middle East, Central, South and Southeast Asia, Oceania and East Africa, where 2.85 billion people are currently at risk of infection and 70-80 million clinical cases are reported each year (Mueller et al. 2009, Guerra et al. 2010). The emergence of drug-resistant strains and severe (sometimes fatal) disease challenges the traditional view of vivax malaria as a benign infection (Price et al. 2009). Recent epidemiological trends in Brazil illustrate the importance of P. vivax as a re-emerging pathogen. The annual incidence of Plasmodium falciparum (the predominant malaria parasite species between 1985-1990) decreased steadily during the 1990s, while that of P. vivax maintained an upward trend. Both P. falciparum and P. vivax are still transmitted across the Amazon Basin, with rare Plasmodium malariae infections. However, P. vivax now causes 85% of the 315,000 malaria cases reported in this country each year (Oliveira-Ferreira et al. 2010), suggesting that this species may be less susceptible to the malaria control strategies currently used in Brazil. Understanding the genetic structure of P. vivax is essential to accurately describing the transmission dynamics of vivax malaria. Population genetics data are crucial, for example, to predict how fast phenotypes of interest, such as novel antigenic variants, particular relapsing patterns or drug resistance, arise and spread in natural populations (Zilversmit & Hartl 2005). The current data on P. vivax population genetics lags behind that of P. falciparum, primarily due to the scarcity of appropriate genetic markers. As we cannot propagate the parasite in continuous culture in vitro (Udomsangpetch et al. 2008), the phenotypic diversity of natural human infections from P. vivax remained unexplored until the early 1990s. Advances in molecular methods, especially implementation of polymerase chain reaction (PCR)-based protocols to amplify parasite DNA, allowed for genotyping of malaria parasite samples obtained directly from patient blood, without a preceding in vitro culture step (Kimura et al. 1990). The relative paucity of genetic markers hampers further investigation into the genotypes that underlie phenotypes such as relapse pattern, virulence and drug resistance. In this review, we broadly classify the available molecular markers into two groups: (i) markers that are clearly under natural selection, that is, markers that map to genome regions associated with adaptive traits and (ii) markers that are neutral or nearly neutral, that is, those that are not obviously influenced by natural selection. The extensive polymorphism found in loci associated with P. vivax adaptive traits, such as those coding for surface antigens and drug-resistance (Cui et al. 2003a), reflects the combined effects of the parasite′s population history and selective constraints imposed by the host′s immunity and drug usage (Escalante et al. 2004). Thus, these loci do not provide much information on the P. vivax population structure. Neutral and nearly neutral markers, in contrast, may allow for unbiased estimates of genetic variation, population structure and gene flow in natural parasite populations. When comparing different markers and parasite typing systems, a clear quantitative definition of genetic diversity is required. One of the most popular methods of summarising genetic diversity levels is to report virtual heterozygosity (HE) values. Virtual HE is the average probability that a pair of alleles randomly obtained from the population is different. Whenever appropriate, in this review we provide HE estimates obtained using different markers for different populations. Molecular markers under selection - Until recently, most genetic markers available for characterising natural P. vivax populations were orthologues of previously identified P. falciparum antigen-coding genes. The well-characterised polymorphic regions in these genes have been extensively used to analyse genetic diversity patterns in P. falciparum (Roy et al. 2008). A similar approach has been explored in vivax-oriented studies (Cui et al. 2003a). The following single-copy genes in P. vivax have often been used in these studies: gam1, coding for the gametocyte antigen 1, csp, coding for the circumsporozoite protein (CSP), msp1 and msp3alpha, coding for the merozoite surface proteins (MSP)-1 and 3α, respectively, ama1, coding for the apical membrane antigen 1, and dbp, coding for the Duffy binding protein (DBP) (Table Ia and Table 1b). A notable feature of several P. vivax surface antigens (including major vaccine-candidate molecules) is tandem arrays of relatively short amino acid motifs. The CSP in P. vivax (PvCSP), an abundant antigen on the surface of sporozoites that has been extensively used as a vaccine development target, has immunodominant B-cell epitopes that map to central repeats between nonrepetitive sequences (Nardin & Zavala 1998). PvCSP displays two major types of nonapeptide repeats [most commonly GDRA(D/A)GQPA and ANGA(G/D)(N/D)QPG], which define the variants known as VK210 and VK247, respectively (Arnot et al. 1985, Rosenberg et al. 1989) (Fig. 1). Both VK210 and VK247 variants are globally distributed, but geographic biases have been described (Cochrane et al. 1990, Qari et al. 1993a, b). Similar repetitive sequences are found in Brazil, Southeast Asia and Papua New Guinea (Qari et al. 1991, 1992). However, markedly divergent sequence variants were found in isolates from China and North Korea (Mann et al. 1994). Although VK210 and VK247-type sequences often occur in sympatric parasite populations, there are no examples of a hybrid repeat array with both repeat types (Lim et al. 2005). A third type of repeat unit [APGANQ(E/G)GAA], identical to that described for Plasmodium simiovale, characterises the so-called P. vivax-like parasites (Qari et al. 1993a). Although P. vivax-like CSP repeats have been found in parasite isolates across the world, its global distribution remains unconfirmed (Gopinath et al. 1994). In Brazil, P. vivax-like parasites have been identified only in mixed-clone infections with VK210-type or VK247-type parasites (Machado & Póvoa 2000). More recently, complex mixtures of parasites harbouring several PvCSP types and co-infecting the same hosts have been demonstrated by Henry-Halldin et al. (2011) in Papua New Guinea. The nonapeptide sequences in PvCSP are repeated ~20 times in the array. Insertions and deletions in the central repeat domain, from either sexual recombination during meiosis or intrahelical strand-slippage events during mitotic DNA replication (McConkey et al. 1990), generate novel CSP variants that may be positively selected if the mutant parasites evade a host's immunity. The central repetitive domain of P. falciparum CSP (PfCSP) contains extensive variation. This domain consists of a variable number of four-mer NANP and NVDP motif copies without breaking down the tight linkage between polymorphic sites in flanking sequences, as in interhelical exchanges during meiosis (Rich et al. 1997). This pattern suggests that these repeats undergo frequent intrahelical recombination that either add or delete repeat units during mitotic DNA replication (Fig. 2), as other short repeats, such as microsatellite and minisatellite-type sequences (Levinson & Gutman 1987). Frequent mitotic recombination coupled with positive selection of new variants might accelerate sequence evolution even where meiotic recombination and outcrossing are relatively uncommon in malaria parasite populations (Rich et al. 2000). In length variation arrays, whether length variation affects B-cell epitope recognition is uncertain, but the conformational nature of repetitive epitopes in PfCSP (Monette et al. 2001) supports this hypothesis. We have recently examined whether similar recombination mechanisms create significant variation in the PvCSP repeat arrays, which consist of longer (9-mer) repetitive motifs (Patil et al. 2010). We investigated patterns of sequence diversity in PvCSP encoding gene alleles in sympatric P. vivax isolates from an area of low malaria endemicity in Brazil. In these isolates, we used single-nucleotide polymorphism typing to examine the haplotype structure of chromosome 8, where the PvCSP encoding gene is located. We sought to determine whether the repetitive domain in the csp locus was significantly diverse under conditions of low malaria endemicity, which reduces effective meiotic recombination rates in local parasites. We confirmed previous findings (Arnot et al. 1985, 1990, Qari et al. 1992, 1994, Mann et al. 1994) of substantial nucleotide sequence diversity in repeat arrays from 45 isolates. They were analysed using nine different csp alleles, each with a unique arrangement of six different nonapeptide repeat sequences, and all were of the VK210 type. Notably, different nucleotide motifs, termed repeat allotypes, coded for the same VK210-type repeat units, indicating that some conservation is maintained at the amino acid level (putatively due to functional constraints), but not necessarily at the nucleotide level (Rich et al. 1997, 2000). Most repeat units in the same csp allele were either identical or nearly identical to each other, consistent with their recent expansion in the repeat array. We found strong linkage disequilibrium at sites across the chromosome 8 segment flanking the csp locus, consistent with rare meiotic recombination in this region. This suggests that repeat array diversity may not be severely constrained by low meiotic recombination rates that are typical in areas with low malaria endemicity. New repeat variants may be readily created by nonhomologous recombination events with potential implications for PvCSP-based vaccine development. The MSP-1 and 3α are additional examples of blood-stage vaccine-candidate antigens with repetitive arrays. The 200-kDa MSP 1 in P. vivax (PvMSP-1), originally described at the molecular level by del Portillo et al. (1991), is a major target of naturally acquired (Nogueira et al. 2006) and vaccine-induced immunity (Yang et al. 1999, Herrera et al. 2007). The protein contains six highly polymorphic domains (4 of them repetitive) flanked by seven fairly conserved sequences (Putaporntip et al. 2002) (Fig. 1). Sequence analysis has confirmed that blocks 1, 3 and 5 in PvMSP-1 are conserved at the protein level, while blocks 2, 4 and 6 are highly variable (Kolakovich et al. 1996, Figtree et al. 2000, Hoffmann et al. 2003, Bastos et al. 2007, Farooq et al. 2009, Kim et al. 2009, Veron et al. 2009, Zakeri et al. 2010). The extensive sequence divergence in variable domains of PvMSP-1 (amino acid similarity, 21-34%) has been maintained by balanced selection, most likely as a result of variant-specific immune pressure (Putaporntip et al. 2006). However, the extent to which PvMSP-1 sequence diversity affects immune recognition for this major malaria-vaccine candidate antigen remains uncertain. To address this topic, we have recently compared naturally acquired antibody responses to three polymorphic domains and the highly conserved PvMSP-1 C-terminal domain in a rural, malaria-exposed Amazonian cohort. We expressed 15 recombinant proteins corresponding to PvMSP-1 variants commonly found in local parasites and showed that less than one-third had detectable IgG antibodies to at least one expressed variant of blocks 2, 6 and 10. However, 54.3% recognised the invariant C-terminal domain PvMSP-119. Although the proportion of PvMSP-1 variant responders increased substantially during subsequent acute P. vivax infections, the specificity of IgG antibodies did not necessarily match the PvMSP-1 variant(s) found in infecting parasites (Bastos et al. 2007). The mechanisms underlying the limited immune recognition of PvMSP-1 variants that are known to circulate in a given endemic area remain unclear. P. vivax evidences an allelic form diversity across the msp1 gene. In contrast, P. falciparum exhibits a clear allelic dimorphism over a long region in the msp1 locus (that is, sequences can be grouped into 1 of 2 major allelic types, MAD20 and K1) (Roy et al. 2008). Frequent meiotic recombination events that shuffle different sequence types to generate new alleles are responsible for the mosaic structure of PvMSP-1 (Putaporntip et al. 1997, 2002). Recently, Han et al. (2011) compared PvMSP-1 alleles from re-emergent P. vivax parasites collected in the Republic of Korea over the past decade. They showed an increase in allelic diversity for variable domains of this protein, suggesting that recombination independently changed protein domains. Moreover, the greater similarity between allelic sequences within P. vivax than between P. vivax and closely related species was taken as evidence that polymorphisms in P. vivax arose recently. This is in stark contrast to the P. falciparum pattern, wherein different alleles are estimated to have diverged more than 25 million years ago (Polley et al. 2005). However, the cause for the large differences in this genetic history for these highly divergent parasites remains obscure. It may reflect either the significantly different sequence structure of the orthologues, different molecular functions for the gene in the two species, a different population history, or another event. Proteolytic processing of PvMSP-1 generates a C-terminal fragment of 42 kDa (PvMSP-142), which is subsequently cleaved to generate two smaller fragments with apparent molecular masses 33 kDa (PvMSP-133) and 19 kDa (PvMSP-119). Only the small, PvMSP-119 fragment remains at the merozoite surface during and after erythrocyte invasion (Holder 2009). Limited polymorphism has been observed in the C-terminal 19-kD PvMSP-1 fragment, with only two polymorphic sites (Soares et al. 1999, Dias et al. 2011). PvMSP-142, like PfMSP-142, exhibits extensive genetic polymorphism in natural infections (Escalante et al. 1998, Conway et al. 2000, Putaporntip et al. 2002, Pacheco et al. 2007, Thakur et al. 2008, Dias et al. 2011). The P. vivax msp3α gene encodes a MSP with an apparent molecular mass ranging from 148-150 kDa. The protein contains an alanine-rich central domain that is predicted to form a coiled-coil tertiary structure (Galinski et al. 1999) (Fig. 1). The gene coding for the MSP-3α (PvMSP-3α) is highly polymorphic and its variability has been assessed by restriction fragment length polymorphism-PCR analysis (Bruce et al. 1999). The suitability of PvMSP-3α as a molecular marker has been confirmed in several geographic parasite populations: Papua New Guinea (Bruce et al. 1999), Thailand (Cui et al. 2003b, Mascorro et al. 2005), Pakistan and Iran (Zakeri et al. 2006, 2010, Khatoon et al. 2010), Venezuela (Ord et al. 2005), Peru (Sutton et al. 2009), Colombia (Cristiano et al. 2008), India (Prajapati et al. 2010) and French Guiana (Veron et al. 2009). However, classifying restriction fragments according to size after electrophoresis is relatively subjective, which hampers large-scale analyses and reduces comparisons between studies. In addition, complex restriction patterns generated by mixed-clone infections may be difficult to interpret. The P. vivax apical membrane antigen-1 (PvAMA-1) is an immunogenic, type 1 integral membrane protein, which is expressed at the apical surface of merozoites and sporozoites. It seems to play a role in erythrocyte and hepatocyte invasion (Hodder et al. 2001, Silvie et al. 2004). A number of studies have addressed polymorphism in that PvAMA-1 encoding gene in various parts of the world, such as Asia, Oceania and South America (Cheng & Saul 1994, Figtree et al. 2000, Han et al. 2002, Chung et al. 2003, Rodrigues et al. 2005, Gunasekera et al. 2007, Rajesh et al. 2007, Grynberg et al. 2008, Thakur et al. 2008, Moon et al. 2010). The PvAMA-1 protein includes three extracellular domains, known as domains I, II and III (Pizarro et al. 2005) (Fig. 1). Most studies show a high degree of genetic variability in domain I (Ord et al. 2008, Thakur et al. 2008, Putaporntip et al. 2009), while the Sri Lanka domain II is the most polymorphic (Gunasekera et al. 2007). A comparison of PvAMA-1 domain I sequences from 320 worldwide isolates revealed a striking divergence between Old World populations and those from Brazil (Grynberg et al. 2008, Putaporntip et al. 2009). Sequence analysis of Sri Lankan isolates suggested that balancing selection maintains polymorphism at the P. vivax ama1 locus (Gunasekera et al. 2007), while no strong evidence for balancing selection was found in Thailand (Putaporntip et al. 2009). A number of studies have reported a substantially lower variability at this locus in P. vivax than in P. falciparum, implying that natural selection acts differently at this locus in the two species (Gunasekera et al. 2007, Ord et al. 2008, Putaporntip et al. 2009). P. vivax DBP (PvDBP) plays an important role in the formation of an irreversible junction between P. vivax merozoites and its receptor, Duffy antigen/receptor for chemokines (DARC), at the surface of immature red blood cells (reticulocytes). This is a key step in host cell invasion (Singh et al. 2005, Galinski & Barnwell 2009). In vitro, antibodies against PvDBP inhibit DBP binding to DARC and block invasion of human erythrocytes (Michon et al. 2000, Grimberg et al. 2007, Ceravolo et al. 2008). The most polymorphic segment of this protein is the erythrocyte-binding motif, a 170 amino-acid stretch located in the cysteine-rich domain II (Ranjan & Chitnis 1999, VanBuskirk et al. 2004, Hans et al. 2005) (Fig. 1-). This segment of the protein has been used as molecular marker in genetic population studies of field isolates from Papua New Guinea (Tsuboi et al. 1994, Xainli et al. 2000), Colombia (Ampudia et al. 1996), South Korea (Kho et al. 2001), Brazil (Sousa et al. 2006, 2010) and Thailand (Gosi et al. 2008). Recently, the importance of selective diversifying pressure has been shown for specific regions of the protein structure (Sousa et al. 2010). Although molecular epidemiological studies using P. vivax gam1 gene have revealed variability in P. vivax isolates from Sri Lanka (Snewin et al. 1995), Korea (Kho et al. 2001) and India (Prajapati et al. 2006), amplification of this gene has been associated with artefacts (Imwong et al. 2001, 2005). For this reason, P. vivax gam1 is no longer used as a reliable genetic marker. Neutral or nearly neutral molecular markers - Feng et al. (2003) described 33 tandem repeat (TR) arrays consisting of repeat units with four or more nucleotides. However, not much diversity was revealed by analysing these arrays in four monkey-adapted P. vivax strains (India VII, Salvador-I, Belem, Thai-NYU). Leclerc et al. (2004) used the eight most polymorphic TR loci described by Feng et al. (2003) to show much more variability in TRs than microsatellites in isolates from different endemic areas. However, little TR diversity was found within populations (Tables II, III). Rezende et al. (2009) selected the five most polymorphic loci from Leclerc's study for their analysis of field isolates from four regions in Brazil. This set of TR markers again revealed relatively little diversity in Brazilian isolates (Table III). Most TR markers had a clearly predominant allele. Moreover, these markers were inefficient in detecting multiple-clone infections and were unable to group the isolates according to their geographical origin (Rezende et al. 2009). As these TR markers map to a 100-kb region in chromosome 8 with many putative protein-coding genes (including csp), natural selection is expected to affect them, even if indirectly, as a result of hitchhiking (Feng et al. 2003). Thus, these loci may not be effectively neutral given their proximity to sequences under strong diversifying or purifying selection (Leclerc et al. 2004). An alternative explanation for the low variability in TR loci may include their structure, as they are relatively short arrays consisting of long (> 6 bp) repeat units. These sequences are less prone to strand slippage events (Fig. 2) than typical microsatellite-type sequences consisting of long arrays of short repeat motifs. The current markers of choice for large-scale population genetic studies in eukaryotes are highly polymorphic and short (1-6 bp-long) TRs, known as microsatellites (Schlötterer 1998). In contrast to the ~1,000 polymorphic microsatellite loci currently available for P. falciparum typing, it was demonstrated that a few dozen microsatellite loci are polymorphic in P. vivax field isolates (Table II). Despite these limitations, microsatellite-based studies of the parasite have provided valuable information on P. vivax population structure and diversity in the Americas, Asia and Africa (Ferreira et al. 2007, Karunaweera et al. 2007, 2008, Koepfli et al. 2009, Orjuela-Sánchez et al. 2009a, Gunawardena et al. 2010, Rezende et al. 2010, Van den Eede et al. 2010a, b, 2011). The first microsatellite described for P. vivax was a dinucleotide (AT) repeat, used to identify high variability in 89 isolates from Papua New Guinea (Gómez et al. 2003) (Tables II, III). Leclerc et al. (2004) reported low variability in dinucleotide microsatellite loci. In this study, only one of 13 microsatellites showed extensive variability and nine were entirely monomorphic among the eight P. vivax populations analysed (Table III). Using a draft of the unpublished P. vivax genome, Imwong et al. (2006) designed primer pairs for 11 dinucleotide microsatellites and showed extensive variability in P. vivax populations from areas with intermediate to high levels of endemicity. They compared their findings with those of Leclerc et al. (2004) and explained the discrepancy in microsatellite diversity levels as the result of repeat array length at the loci studied. They suggested that microsatellite diversity correlates positively repeat array length, as the rate of strand-slippage events that create diversity increases exponentially with repeat array lengths. Recently, Rezende et al. (2010) corroborated this hypothesis using a set of microsatellites with di, tri and tetra nucleotide repeat units. They highlighted the importance of microsatellite loci structure in genetic diversity studies using malaria parasites. Karunaweera et al. (2007) described a new set of highly polymorphic tricleotide and tetranucleotide microsatellites, which are expected to yield more accurate allele scores than dinucleotide markers (Anderson et al. 1999). P. vivax genome sequencing allowed for further characterisation of a panel including ~160 microsatellites (Carlton et al. 2008). While searching for a repeat unit with 2-6 nucleotides, the authors found only one microsatellite locus with repeats consisting of more than four nucleotides. Most (76%) loci had perfect repeat units (Fig. 3A), in contrast with the markers described by Karunaweera et al. (2007). All except one microsatellite were assigned to chromosomes, but their chromosome distribution was clearly heterogeneous. Chromosomes 1 and 4 had the lowest marker density, whereas chromosomes 14 and 9 had the highest density (Fig. 3B). Despite the relatively low AT content in the P. vivax genome, when compared with that of P. falciparum, P. vivax microsatellite-type repeats are particularly AT-rich (Fig. 3C). To characterise polymorphic microsatellite markers, these loci were analysed in eight monkey-adapted P. vivax strains (Brazil I, Miami II, Pakchong, Panama I, Nicaragua, Thai II, Vietnam IV and Indonesia XIX). Nineteen markers were monomorphic among these samples, none were polymorphic in all strains and the large majority of markers were polymorphic only in 2-4 strains (Fig. 3D). Further, only one-fourth of the markers shown to be polymorphic in natural P. vivax populations were also identified as polymorphic using these eight P. vivax strains. High variability in P. vivax isolates has been demonstrated using different sets of microsatellites (Table III). However, this comparison is complicated, as it is difficult to determine how much of the variation depends on the set of markers and how much is due to genetic diversity in the studied populations (Imwong et al. 2007a, Koepfli et al. 2009). Currently, the most widely used markers are a set of 14 polymorphic microsatellites described by Karunaweera et al. (2007), which were used to analyse the population structures of P. vivax in Brazil, Vietnam, Sri Lanka, Myanmar, Ethiopia and Peru (Ferreira et al. 2007, Karunaweera et al. 2008, Gunawardena et al. 2010, Van den Eede et al. 2010a, b, 2011). Using the same set of markers, similar variability was detected in different geographical areas, with HE ranging from 0.71 in Brazil to 0.86 in Myanmar and Vietnam; only Peru showed a slightly lower variability (HE = 0.58). More recently, Joy et al. (2008) selected 24 microsatellites of ~160, described by Carlton et al. (2008), and used them to show local adaptation between mosquito vectors and different genetic populations of P. vivax from Mexico. There was no correlation between the number of alleles identified by Carlton et al. (2008) and the variability identified using a large number of P. vivax field isolates using the same set of markers by Joy et al. (2008). These data reinforce the uncertainty in using laboratory strains to identify polymorphic markers (Fig. 4). Koepfli et al. (2009) showed high variability in Papua New Guinea isolates using this set of microsatellites and three selected from other studies (Table II). Rezende et al. (2010) described a set of 11 microsatellites used to show a positive correlation between transmission levels and genetic variability in P. vivax isolates from geographical regions in Brazil. Taken together, these data suggest that, regardless of endemicity in the isolates' geographical origin, most studies show that P. vivax populations are highly variable across the globe. Practical uses for P. vivax genotyping - The vast field of infectious disease molecular epidemiology has produced a number of examples involving practical applications for molecular markers in addressing issues of clear biological and public health importance. Here, we briefly review four areas of potential interest: (i) detection of naturally occurring multiple-clone infections, (ii) after-treatment follow-up in clinical trials, (iii) between-species comparisons of genetic diversity levels and (iv) tracking of the geographic origin of infections. Over the last two decades, molecular markers have revealed that many natural P. vivax infections comprise a complex mixture of clonal populations; that is, multiple-clone vivax infections are fairly common in human hosts (Havryliuk & Ferreira 2009). The earliest attempts to investigate the clonal diversity of P. vivax isolates in human infections focused on characterisation of phenotypes using monoclonal antibodies or isoenzymes (Udagama et al. 1987, 1990, Joshi et al. 1989), an approach that required blood samples with relatively high parasitaemias. As a rule, multilocus analysis, such as microsatellite typing, detects more multiple-clone infections than single-locus analysis, and augmenting the number of markers increases the chance of detecting multiple clones. For example, in 90 P. vivax isolates from Thailand, Cui et al. (2003b) found a multiple-clone infection rate of 25.6% using the PvCSP encoding gene alone, 19.3% using PvMSP-3α alone and 35.6% when the markers were combined. Particularly surprising is the extensive clonal diversity of P. vivax infections in areas with relatively low malaria transmission, such as Brazil, Colombia and Sri Lanka (Ferreira et al. 2007, Imwong et al. 2007b, Karunaweera et al. 2008, Orjuela-Sánchez et al. 2009a, Gunawardena et al. 2010). The evolutionary and epidemiological consequences of multiple-clone P. vivax infections have not been well-investigated. However, experimental rodent malaria models suggest that, for two or more genetically distinct clones in the same host, intra-host competition for limited resources may select for P. vivax traits representing major public health challenges, such as increased virulence, transmissibility and antimalarial drug resistance (reviewed by Havryliuk & Ferreira 2009). Molecular markers have also been used to type P. vivax infections after drug treatment. Reappearance of parasitaemia after drug treatment can result from either recrudescence of surviving asexual blood-stage parasites; relapse from dormant liver stages, known as hypnozoites; or new infections with unrelated parasites. Paired parasite molecular genotyping distinguishes between recrudescences (with the same genotype as the initial infection) and new infections (with a different genotype). Until recently, relapses were thought to be caused by hypnozoites that are genetically identical to the blood-stage parasites found in primary infections (Craig & Kain 1996, Kirchgatter & del Portillo 1998). This suggested that molecular methods could easily discriminate relapses, which would have the same genotype as the primary infection, from new infections, which would have a different genotype. This view has been challenged by the recent discovery of different parasite genotypes in primary infections and relapses for 72% of P. vivax-infected patients from Thailand, India and Myanmar (Imwong et al. 2007b). Accurate detection of multiple-clone P. vivax infections is even more important in light of this report. If the primary infection comprises multiple clones, then some of them may have been missed or partially characterised or, even worse, had their alleles combined to create artificial haplotypes during genotyping. Likewise, the relapsing clone could have been either missed or incorrectly typed during the primary infection, leading to a false conclusion that different genotypes were present during the primary and relapse infections. The current consensus is that relapses may originate from reactivation of either the same parasite clone found in the primary bloodstream infection (homologous hypnozoites) or another, genetically different clone (heterologous hypnozoites). Our recent microsatellite analysis of 28 paired acute-infection and recurrence parasites from rural Amazonia revealed only two pairs of identical haplotypes (consistent with recrudescences or reactivation of homologous hypnozoites) and four pairs of related haplotypes (sharing alleles at 11-13 of 14 microsatellites analysed) (Orjuela-Sánchez et al. 2009a). As the characteristics of endemic settings may influence the genetic diversity of the parasite, noteworthy findings could be obtained from sympatric P. vivax and P. falciparum isolates that are analysed using similar protocols. Two comparisons of microsatellite diversity in human malaria parasite species co-circulating in the same area in Brazil have been published. Both suggest that P. vivax infections are more diverse (that is, they comprise a higher number of alleles and virtual HE) and comprise multiple clones more often than P. falciparum infections in rural Amazonia (Ferreira et al. 2007, Orjuela-Sánchez et al. 2009b). As P. vivax currently predominates in Brazil, differences in species-specific transmission levels might translate into differences in genetic diversity. However, we recently found more microsatellite diversity in P. vivax compared with P. falciparum populations in western Cambodia, where both species are similarly prevalent (MU Ferreira and RM Fairhurst, unpublished observations). Thus, we suggest that the higher microsatellite diversity found in P. vivax isolates may reflect greater plasticity in microsatellite-type short repeats for this species. Finally, standardised molecular methods applied to worldwide populations may also help characterise population or region-specific molecular barcodes. This is essential for tracking the geographic origin of infections. A microsatellite-based analysis of P. vivax samples from Sri Lanka, Ethiopia and Myanmar recently showed that parasites may be classified as originating from Asia or Africa with 70-80% accuracy. This suggests that microsatellite data might be useful in predicting the origin of P. vivax parasites (Gunawardena et al. 2010). Further analyses are required to confirm whether microsatellite-based molecular barcodes are useful for determining whether particular infections were either transmitted locally or imported into areas approaching the elimination phase. References
Copyright © 2011 - Instituto Oswaldo Cruz - Fiocruz The following images related to this document are available:Photo images[oc11136t3.jpg] [oc11136f4.jpg] [oc11136t1b.jpg] [oc11136f1.jpg] [oc11136f2.jpg] [oc11136f3.jpg] [oc11136t1a.jpg] [oc11136t2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}