 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
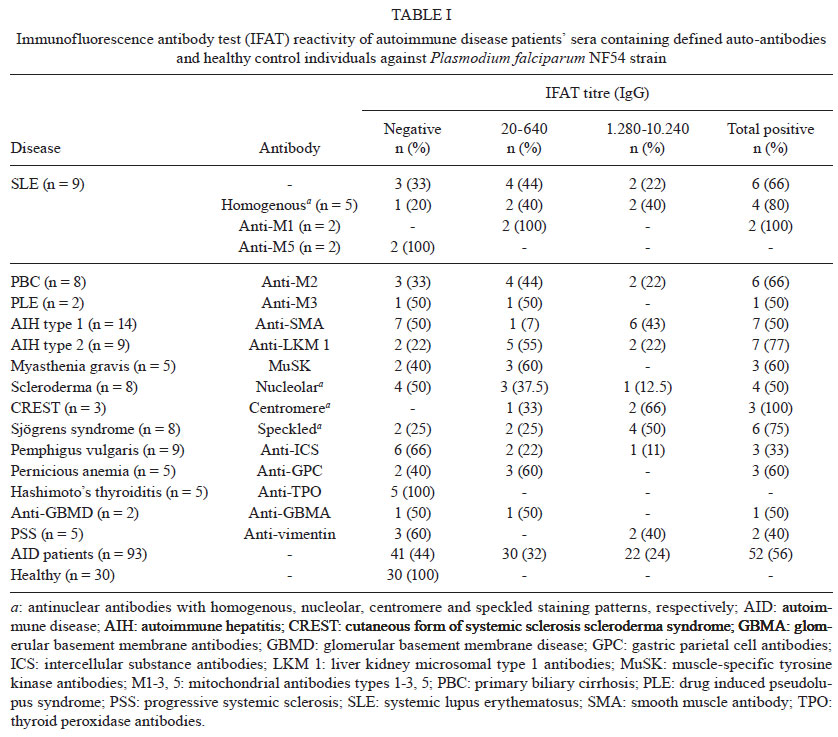
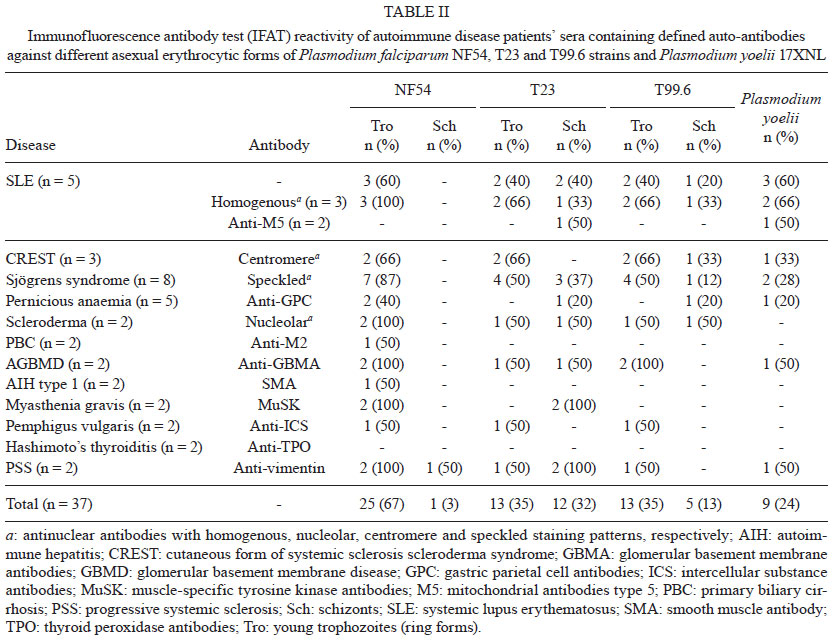
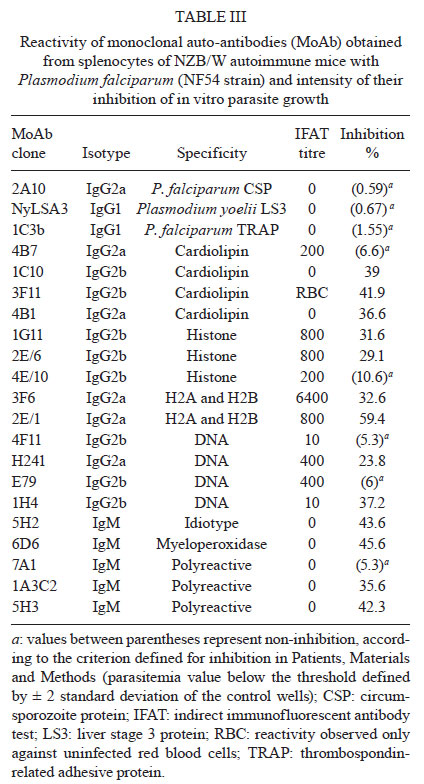
Memórias do Instituto Oswaldo Cruz, Vol. 106, Special Issue, pp. 44-51 Original Article Monoclonal auto-antibodies and sera of autoimmune patients react with Plasmodium falciparum and inhibit its in vitro growth Karima BrahimiI; Yuri Chaves MartinsII; Graziela Maria ZaniniII; Maria de Fátima Ferreira-da-CruzII; Cláudio Tadeu Daniel-RibeiroII, + ILaboratorie
de Parasitologie Biomédicale, Institut Pasteur, Paris, France Received 12 December
2010 Code Number: oc11139 Abstract The relationship between autoimmunity and malaria is not well understood. To determine whether autoimmune responses have a protective role during malaria, we studied the pattern of reactivity to plasmodial antigens of sera from 93 patients with 14 different autoimmune diseases (AID) who were not previously exposed to malaria. Sera from patients with 13 different AID reacted against Plasmodium falciparum by indirect fluorescent antibody test with frequencies varying from 33-100%. In addition, sera from 37 AID patients were tested for reactivity against Plasmodium yoelii 17XNL and the asexual blood stage forms of three different P. falciparum strains. In general, the frequency of reactive sera was higher against young trophozoites than schizonts (p < 0.05 for 2 strains), indicating that the antigenic determinants targeted by the tested AID sera might be more highly expressed by the former stage. The ability of monoclonal auto-antibodies (auto-Ab) to inhibit P. falciparum growth in vitro was also tested. Thirteen of the 18 monoclonal auto-Ab tested (72%), but none of the control monoclonal antibodies, inhibited parasite growth, in some cases by greater than 40%. We conclude that autoimmune responses mediated by auto-Ab may present anti-plasmodial activity. Key words: auto-antibodies - autoimmune diseases - autoimmunity - immune protection - malaria - Plasmodium falciparum Introduction Malaria and autoimmunity are frequently present together, although the relationship between them is poorly understood. Malaria may be associated with autoimmunity by three different ways. The first of them was hypothesised by Greenwood (1968) who considered that malaria infection could protect against the development of autoimmune diseases (AID). This hypothesis has been fundamentally supported by epidemiological records showing that AID, such as systemic lupus erythematosus (SLE), sarcoidosis and multiple sclerosis, are less frequent in populations exposed to malaria (Greenwood 1968, Zoutendyk 1970, Butcher 1995, Sotgiu et al. 2008). Furthermore, experimental data have shown that Plasmodium berghei infection can prevent or delay the onset of disease in murine models of spontaneous AID development (Greenwood et al. 1970, Hentati et al. 1994, Butcher 2008). A second type of association between malaria and autoimmunity is the classical assumption that plasmodial infection induces autoimmune reactions in the host that are related to the genesis of specific disease complications, such as anaemia (Zuckerman 1964, Lefrançois et al. 1981, Banic et al. 1991), nephritis (Burchard et al. 2003, Elsheikha & Sheashaa 2007), thrombocytopenia (SØrensen et al. 1984) and, more recently, cerebral malaria (Lang et al. 2005, Basal et al. 2009). The third hypothesised relationship, which is the least predictable and the most controversial, is that autoimmunity could participate in the development of anti-malarial immunity. This was first proposed by Jayawardena et al. (1979), who considered that part of the IgM response occurring during malaria infections could be constituted by protective auto-antibodies (auto-Ab) directed against modified determinants exposed at the erythrocyte membrane following parasite infection. Later, Jarra (1983) postulated that the acquisition of immunity against blood stage parasites would not be possible without the development of a simultaneous autoimmune response against erythrocytes, even if this resulted in immunopathology. Our research team has since expanded on this theory (Daniel-Ribeiro 2000). Indeed, auto-Ab with various specificities, such as double and single-stranded DNA (Daniel-Ribeiro et al. 1984c, Zouali et al. 1986, Zanini et al. 2009), erythrocytes (Lefrançois et al. 1981, Vivas et al. 2008), lymphocytes (de Souza & Playfair 1983), phospholipids (Bate et al. 1992, Vivas et al. 2008), ribonucleoprotein (Zouali et al. 1986, Daniel-Ribeiro et al. 1991, Zanini et al. 2009), RNA (Kreier & Dilley 1969), smooth muscle (Quakyi et al. 1979, Poels et al. 1980, Daniel-Ribeiro et al. 1991), cardiolipin (Consigny et al. 2002) and cytosol proteins (Dugué et al. 2004) are evident in the course of malaria infection. It seems that the specificities of the auto-Ab produced differ according to the degree of anti-parasite immunity (Poels et al. 1980) and that they are not related to organ-specific, partially sequestered auto-antigens, such as thyroglobulin (Daniel-Ribeiro 2000). In addition, it has been demonstrated that sera from SLE patients can react with plasmodial antigens and inhibit the in vitro growth of Plasmodium falciparum (Singh et al. 2001, Zanini et al. 2009). Taken together, these data suggest that autoimmune responses confer anti-parasite activity, which can be mediated through auto-Ab and/or serum factors that may cooperate with the host immune response during plasmodial infections. The aim of the present work was to further address the relationship between autoimmunity and anti-malarial responses by determining the presence and pattern of reactivity to plasmodial antigens of sera from AID patients who were not previously exposed to malaria and the effect of monoclonal auto-Ab with different specificities on the in vitro growth of P. falciparum. Patients, Materials and Methods Samples - Sera from 93 AID patients, who had not been previously exposed to malaria, were kindly donated from the late professor Jean-Claude Homberg (Laboratories of Immunology and Haematology of Saint Antoine Hospital, Paris, France). All patients were clinically evaluated and diagnosed as experiencing an AID. The patients were selected because they had only one defined auto-Ab among the usually ones characteristics of each disease. For example, the SLE patients whose serum contained homogeneous anti-nuclear antibodies did not have detectable anti-DNA, -Smith, -RNP, -SSA, -SSB, -histone, -phospholipid, -erythrocyte, -platelet, -neuronal or -mitochondrial antibodies, which can also be found in SLE patients. Sera presented auto-Ab titres ranging from 1:80-1:1,000. A summary of the profile of patients with each AID and their corresponding auto-Ab is shown in Table I. Control serum samples were obtained from 30 clinically healthy individuals from the Laboratory of Malaria Research at Oswaldo Cruz Institute, Oswaldo Cruz Foundation (Fiocruz), Rio de Janeiro, who reported no previous episodes of malaria infection. All patients provided informed consent. All procedures were reviewed and approved by the Fiocruz Research Ethics Committee (0018.1.011.000-03) and were conducted in accordance with the Helsinki Declaration of 1975, as revised in 1983. Indirect immunofluorescent antibody test (IFAT) for detecting reactivity against P. falciparum blood stages - IFAT was performed as previously described (Zanini et al. 2009). Briefly, slides containing 20 asynchronously cultured, P. falciparum-parasitised red blood cells (pRBC) (NF54 strain) per microscopic field (Trager & Jensen 1976), were incubated with sera that was serially diluted in phosphate-buffered saline (PBS), followed by the addition of fluorescein isothiocyanate (FITC)-conjugated goat anti-human IgG (Sigma, St. Louis, MO, USA) diluted in PBS-Evans Blue solution for 30 min at 37ºC. Slides were mounted and analysed using a Zeiss fluorescence microscope. A reciprocal titre of 20 or greater was considered positive. Sera from 37 AID patients were also tested against Plasmodium yoelii 17XNL strain and specific asexual blood stage forms of P. falciparum NF54 (unknown origin), T23 and T99.6 (Thai origin) strains (Table II). For this analysis, the parasite asexual blood stage cycle was synchronised by successive sorbitol treatments (Lambros & Vanderberg 1979) and slides were made when the P. falciparum were in the young trophozoite (ring form) or schizont stages. P. yoelii 17XNL slides were obtained from a Balb/c (Charles River, France) mouse that had been infected intra-peritoneally with 1 x 105 pRBC four days earlier. Parasite culture and inhibition assay - P. falciparum NF54 strain was cultured in Roswell Park Memorial Institute (RPMI) supplemented with 10% human serum as previously described (Butcher 2008). P. falciparum growth inhibition assays were performed in duplicate in 96-well tissue culture plates (NUNC, Denmark). The assay was started with synchronised schizonts (0.5% initial parasitaemia and 2.5% haematocrit) in RPMI medium supplemented with 10% Albumax. Eighteen murine monoclonal auto-Ab with different specificities (Table III), obtained from splenocytes of NZB/W autoimmune mice (Charles River, France) (a kind gift from Professor François Tron, Laboratoire Central d'Immunologie, CHU de Rouen, France) and sera of healthy donors were serially diluted in PBS and tested at a 1/10 final volume. Slides were washed three times in PBS (pH 7.4) for 10 min and incubated with either FITC-anti-mouse IgG (H+L) (Alexa, Interchim, France) or FITC-anti-mouse IgM (Cappel) diluted in PBS-Evans Blue solution for 30 min at 37ºC. The following control monoclonal antibodies (MoAb) were also used: (i) MoAb 2A10 (IgG2a), a stage and species-specific antibody directed to the P. falciparum circumsporozoite protein, which is expressed on the surface of sporozoites but not during the asexual erythrocytic stages (Wirtz et al. 1987), (ii) MoAb Navy yoelii liver stage 3 (NYLS3, IgG1) directed against the 17 kDa liver stage antigen 3 protein of P. yoelii, which is expressed by liver stage parasites within 6 h of sporozoite invasion of mouse hepatocytes, throughout the asexual erythrocytic stages and on merozoites [NYLS3 is species-specific, does not cross-react with blood stage antigens and, as expected, does not inhibit P. falcipa-rum growth (Charoenvit et al. 1995)], (iii) MoAb 1C3 (IgG1, kappa) specific to the thrombospondin-related adhesive protein, which is expressed on both sporozoites and erythrocytic stages of P. falciparum (Robson et al. 1995). It has been previously shown that 1C3 does not inhibit P. falciparum growth (Robson et al. 1995). The endpoint parasitaemia was determined in thin smears from each well by microscopic examination after 72 h of culture at 37ºC, 5% CO2. The inhibitory effect was determined as follows: the mean parasitaemia of five control wells (containing only parasitised erythrocytes and culture medium) was established as 100% of parasite growth. Parasitaemia levels falling within the mean [± 2 standard deviations (SD)] were considered to be within the normal growth range. Sera causing parasitaemia to decrease below the mean by 2 SD were considered inhibitory. The intensity of inhibition was measured by the percent decrease in parasitaemia relative to the mean control value. Statistical analysis - Fisher's exact test was used to evaluate associations between serum reactivity with plasmodial antigens and the presence of AID and auto-Ab. The Chi-square goodness of fit test was used to evaluate whether the observed reactivity frequencies in the subpopulations of patients with different AID differed from the reactivity frequencies found throughout the entire population of AID patients. McNemar's test was used to evaluate if the frequencies of reactive sera against P. yoelii 17XNL and specific asexual blood stage forms of P. falciparum strains were different. Results Reactivity of AID patients' sera with P. falciparum NF54 strain - AID patients presented a high frequency (56%) of sera that reacted with plasmodial antigens when compared with healthy controls (0%) (Table I), as determined by IFAT. With the exception of patients with the limited cutaneous form of systemic sclerosis scleroderma syndrome (CREST) or Hashimoto's thyroiditis, there were no statistically significant relationships in the frequencies of IFAT-positive sera between subpopulations of patients with different AID and the population of AID patients as a whole (p > 0.05 for all comparisons). However, patients with autoimmune hepatitis type 1 presented a higher frequency of IFAT-positive sera with high titres (1,280-10,240) when compared with the sera of patients with other AID (p < 0.05). All three CREST syndrome patients' sera tested positive in IFAT with titres ranging from 400-3,200, and all five Hashimoto's thyroiditis patients' sera were negative. As expected, the reactivity frequency of SLE patients' sera varied with the auto-Ab it contained; however, this difference was not statistically significant, probably due to the small number of patients in each group (p > 0.05) (Table I). Reactivity of AID patients' sera with different asexual blood stage forms of P. falciparum NF54, T23 and T99.6 strains and P. yoelii 17XNL - With the exception of Hashimoto's thyroiditis patients, at least one sample from all AID groups was reactive against the three P. falciparum strains tested (Table II). The frequency of sera reacting against young trophozoites was higher than the frequency of sera reacting against schizonts for all three strains (Table II). However, the reactivity frequencies against the two blood stages were significantly different only for the NF54 and T99.6 strains (p < 0.001 and p < 0.05, respectively). The frequency of reactive sera against the NF54 strain (67%) was higher than those of T23 (50%) or T99.6 (42%) strains (p < 0.05 and p < 0.01, respectively). Nevertheless, there was no statistically significant difference between the frequencies of reactive sera against the T23 and T99.6 strains. The frequency of reactive sera against P. yoelii 17 XNL (24%) was lower than the frequency of reactive sera against any P. falciparum strain (all p < 0.05). Again, the reactivity pattern of SLE patients' sera varied with the associated antibody (Table II). Inhibition of in vitro P. falciparum growth by murine monoclonal auto-Ab - Murine monoclonal auto-Ab were tested for reactivity to P. falciparum antigens using IFAT (Table III). Among the 13 IgG2a or IgG2b isotype clones, 85% tested positive, with titres ranging from 10-6,400. The five clones expressing IgM isotypes gave negative results. One monoclonal antibody (clone 3F11) reacted only against normal RBCs, but all other auto-Abs were reactive against pRBCs. Thirteen of the 18 MoAb (72%) inhibited parasite growth, according to the aforementioned criteria (Table III). Interestingly, inhibition did not correlate with the intensity of reactivity against the parasite, as assessed by IFAT. Even antibodies showing low reactivity (especially those of the IgM isotype) inhibited growth. None of the control MoAb directed against non-expressed antigens inhibited the parasites' in vitro growth. Discussion The results presented here allow us to conclude that (i) sera from patients with different AID can react with plasmodial antigens, (ii) the mammalian antigenic determinants that are recognised by the AID patients' sera studied are generally expressed at higher levels by the trophozoites compared to schizonts, with at least some of the antigens expressed by both P. falciparum and P. yoelii, and (iii) monoclonal auto-Ab with different specificities can inhibit the in vitro growth of P. falciparum. Although previous studies have shown that plasmodium infections induce autoimmune responses and may influence the development of AID [for review, see Daniel-Ribeiro and Zanini (2000)], the potentially protective role of autoimmune responses against plasmodial infections has been more difficult to assess. It has previously been shown that the immune response against phosphatidylinositol and P. falciparum band three neo-antigen, a host derived protein that integrates the "knob" protein complex, correlates with protective immunity (Facer & Agiostratidou 1994, Hogh et al. 1994). Additionally, murine studies have shown that pre-existing haemolysis, generated by treatment with phenylhydrazine or immunisation with phosphatidylcholine, induces partial protection against Plasmodium chabaudi infection (Jarra 1983, Bordmann et al. 1998) and that there is a relationship between the induced anti-erythrocytic auto-Ab titres and protection against P. berghei infection (Schetters et al. 1989). However, the strongest evidence supporting the protective role of autoimmunity in plasmodial infection has been provided by two independent studies, which showed that sera and IgG antibodies from SLE patients can react with and inhibit the in vitro growth of P. falciparum (Singh et al. 2001, Zanini et al. 2009). The present paper expands these findings by showing that sera from patients with other AID, containing auto-Ab of different specificities, can also react against P. falciparum and inhibit the parasite's in vitro growth. We did not test the reactivity of the AID patients' sera against a panel of plasmodia and human cell components to search for patterns of cross-reactivity. However, it is well documented in the literature that sera from patients with different AID cross-reacts with xenobiotics (Selmi et al. 2009) and pathogens, including virus, bacteria and parasites (Capron et al. 1965, Dissous et al. 1986, Daniel-Ribeiro & Zanini 2000, Gregorio et al. 2003, Amano et al. 2005, Blank et al. 2007, Bogdanos & Vergani 2009). This cross-reactivity is the basis for the "molecular mimicry" hypothesis, originally formulated by Damian (1964), who suggested that antigenic determinants of micro-organisms may resemble antigenic determinants of their host. Pathogens use this similarity to avoid the immune response and persist in immunologically competent hosts. Therefore, molecular mimicry between human and P. falciparum antigens could explain the high frequency of reactive AID patients' sera we found. Furthermore, auto-Abs found during the course of AID can also cross-react with different self-structures in the host. For example, anti-DNA auto-Ab from SLE patients can bind to α-laminin, α-actinin, myosin and the NR2 glutamate receptor (DeGiorgio et al. 2001, Mageed & Zack 2002), and the "same" anti-DNA auto-Ab from primary biliary cirrhosis patients can cross-react with mitochondrial antigens (Shimoda et al. 2003). In the same way, anti-cardiolipin (anti-M1) antibodies can bind to phospholipids and β2 glycoprotein I (Harris & Pierangeli 1994). This self-cross-reactive phenomenon could also contribute to our findings. Although we did not ascertain that the auto-Ab was responsible for the reactivity in IFAT, as other auto-Ab frequently present in each AID were not detected during the patients evaluation, and all healthy patients sera were negative for the presence of these pathogenic auto-Ab, our data suggest that this hypothesis is true. Changes in the frequency and titres of reactive sera according to the presence of different auto-Ab in the subpopulation of SLE patients also corroborate this hypothesis. Anti-nuclear, anti-smooth muscle and anti-M1 antibodies are induced during the course of malaria infection and these may be related to the degree of immunity [for review, see Daniel-Ribeiro and Zanini (2000) and Consigny et al. (2002)]. The present work supports these data by showing that sera containing these auto-Abs can react against P. falciparum and that anti-M1 MoAb can inhibit in vitro parasite growth. Interestingly, sera containing anti-smooth muscle auto-Ab had high anti-parasite antibody titres, revealing that these auto-Abs may strongly cross-react against P. falciparum in vivo. As these auto-Abs are associated with acute malaria infections (Poels et al. 1980, Phanuphak et al. 1983, Daniel-Ribeiro et al. 1991), our data may indicate that they are important during the early stages of the anti-parasite immune response. Sera from patients with Hashimoto's thyroiditis did not react with any of the three strains of P. falciparum or P. yoelii 17XNL. This finding agrees with previous works showing that anti-thyroid peroxidise antibodies and anti-thyroglobulin antibodies are absent during human or murine malaria infections (Daniel-Ribeiro et al. 1983, 1984a). To our knowledge, our study is the first to indicate that anti-vimentin, anti-glomerular basement membrane, anti-gastric parietal cell, anti-intercellular substance, muscle-specific tyrosine kinase and anti-liver kidney microsomal type 1 auto-Ab can react against plasmodial antigens. Polyclonal B cells (B2 cells) and auto-antibody-producing B cells (B1 cells) activation, parasite and host antigen cross-reactivity (molecular mimicry) and the presence of altered self-antigens have all been proposed as explanations for the formation of auto-Ab during the course of malaria infection (Abu-Shakra & Shoenfeld 1991, Daniel-Ribeiro & Zanini 2000, Yoder & Goodrum 2001, Kanda et al. 2010). Studies with murine models show thymic atrophy and a decrease in the number of "conventional" thymic T lymphocytes, as well as proliferation and activation of autoreactive extrathymic T cells (mainly NK1.1-TCRint cells) during malaria infection (Mannoor et al. 2001, 2002, Weerasinghe et al. 2001). NK1.1-TCRint cell activation is associated with the proliferation of auto-antibody-producing B1 cells and protection against the infection (Yoder & Goodrum 2001, Kanda et al. 2010). Because a similar mechanism has been shown to occur in certain murine AID states (Miyakawa et al. 2002, Morshed et al. 2002, Kawabe et al. 2004) and because there is an increase of unconventional T cells with NK markers in malaria patients' blood (Watanabe et al. 2003), the production of auto-Ab during the course of plasmodia infections and some AID could overlap with pathways that are necessary for the development of malarial protection. The development of auto-Ab during malaria could be elicited by the release of parasitic, self and altered-self-antigens into the bloodstream due to the destruction of pRBCs during the erythrocytic cycle [for a review, see Daniel-Ribeiro (2000)]. However, if this was true, one would expect the frequencies of reactive sera against schizonts to be the same, or even higher, than that against the young trophozoites, which was not the case. In fact, the AID patients' sera we studied (containing auto-Ab usually expressed during the course of malaria infection) reacted more frequently with the young asexual blood stage forms of P. falciparum than with schizonts. It may be noteworthy that sera from patients with SLE, progressive systemic sclerosis, anti-glomerular basement membrane disease, pernicious anaemia, Sjögren's syndrome or CREST syndrome can also react against P. yoelii 17XNL, suggesting that reactive sera might be specific for plasmodial structures conserved in different species during evolution. Finally, a number of monoclonal auto-Ab derived from transformed lymphocytes of NZB x NZW mice was able to inhibit P. falciparum growth, in some cases by more than 40%. As previous works used only purified IgG preparations (Singh et al. 2001, Zanini et al. 2009), the present study is the first to show that monoclonal auto-Ab can inhibit in vitro P. falciparum growth. This result was expected, because monoclonal antibody cross-reactions between plasmodial and mammalian cells were reported more than two decades ago (Daniel-Ribeiro et al. 1984b, Li et al. 1995, Crandall et al. 1996). The lack of association between IFAT titres and the degree of growth inhibition remains to be explained, but this could be related to the small number of tested samples. However, our control MoAb results indicate that the inhibition results from a specific effect of the Mo-autoAb. We propose two mechanisms by which Mo-autoAb could inhibit plasmodium growth. First, the binding of Mo-autoAb onto erythrocyte membrane receptors could block merozoite-ligands recognition and impair the RBC invasion process. This mechanism would explain why the 3F11 MoAb clone, which had reactivity only against uninfected RBC, inhibited parasite growth. Second, Mo-autoAb may induce parasite or erythrocyte death. Eryptosis is a suicidal erythrocyte death process similar to apoptosis, which can be triggered by various stimuli including anti-blood group A IgG antibodies (Attanasio et al. 2007) and it has recently been shown that P. falciparum erythrocytes die by an autophagic-like cell death under antimalarial pressure (Totino et al. 2008). In that way, Mo-autoAb could react with components of the pRBC or parasite membrane and cause metabolic disturbances, for example an increase in the cytosolic Ca+2 concentration, triggering eryptosis or parasite cell death and inhibiting parasite growth. In summary, our data reinforce the notion that serum from autoimmune patients has anti-plasmodial activity that is mediated through auto-Ab of various specificities. Although we did not rule out the contribution of other serum factors, which should be investigated further for a better understanding of the phenomenon, the data presented here may have important implications on the understanding of the relationship between autoimmunity and malaria. References
Copyright © 2011 - Instituto Oswaldo Cruz - Fiocruz The following images related to this document are available:Photo images[oc11139t1.jpg] [oc11139t2.jpg] [oc11139t3.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}