 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Chromatin regulation in schistosomes and histone modifying enzymes as drug targets Raymond John Pierce*; Florence Dubois-Abdesselem; Stéphanie Caby; Jacques Trolet; Julien Lancelot; Frédérik Oger; Nicolas Bertheaume; Emmanuel Roger Institut Pasteur
de Lille, CIIL, F-59019 Lille, France, Inserm, U 1019, F-59019 Lille, France,
Univ. Lille Nord de France, F-59000 Lille, France, CNRS, UMR 8204, F-59021 Lille,
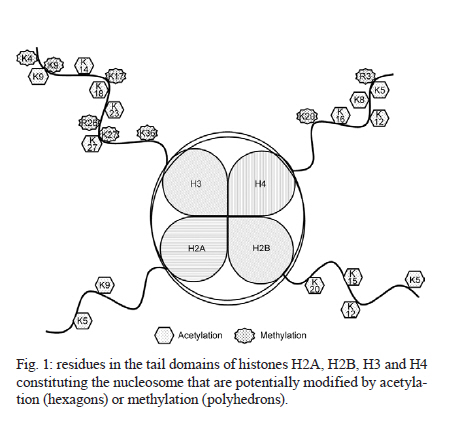
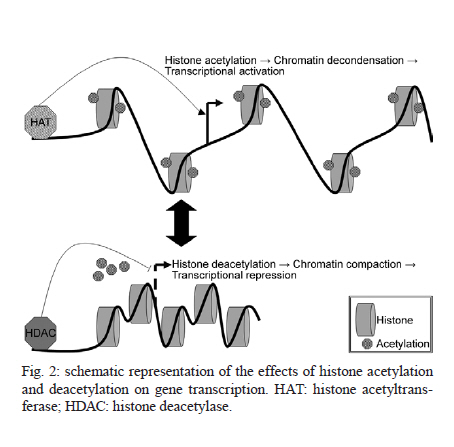
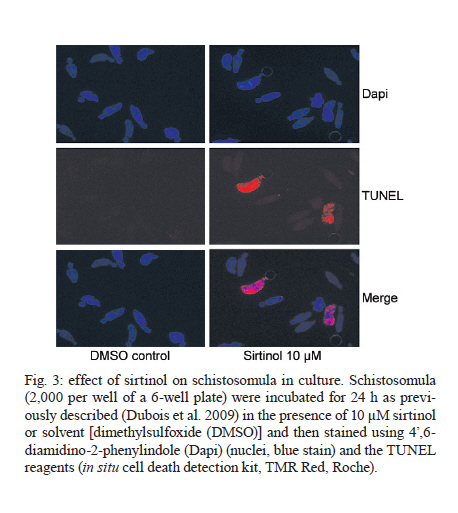
France Code Number: oc11165 ABSTRACT Only one drug is currently available for the treatment and control of schistosomiasis and the increasing risk of selecting strains of schistosome that are resistant to praziquantel means that the development of new drugs is urgent. With this objective we have chosen to target the enzymes modifying histones and in particular the histone acetyltransferases and histone deacetylases (HDAC). Inhibitors of HDACs (HDACi) are under intense study as potential anti-cancer drugs and act via the induction of cell cycle arrest and/or apoptosis. Schistosomes like other parasites can be considered as similar to tumours in that they maintain an intense metabolic activity and rate of cell division that is outside the control of the host. We have shown that HDACi can induce apoptosis and death of schistosomes maintained in culture and have set up a consortium (Schistosome Epigenetics: Targets, Regulation, New Drugs) funded by the European Commission with the aim of developing inhibitors specific for schistosome histone modifying enzymes as novel lead compounds for drug development. Key words: Schistosoma mansoni - histone modifying enzyme - histone deacetylase - inhibitor - drug target The need for new drugs to treat schistosomiasis - The treatment and control of schistosomiasis currently relies on a single drug, praziquantel. Although safe, effective and relatively cheap, the massive use of this drug, for example in the Schistosomiasis Control Initiative in Africa (Fenwick et al. 2009), renders the selection of field strains of schistosome that are resistant to praziquantel more and more likely. Resistant isolates have already been characterized in endemic areas (Doenhoff et al. 2002) and the selection of parasites in the laboratory that are stably resistant in the absence of drug pressure has been demonstrated (Fallon & Doenhoff 1994, Couto et al. 2011). In view of the lag time between the discovery of a new lead compound and its eventual approval for use, it is urgent to start the search early and indeed a number of strategies are being pursued. The challenge is to develop a new drug that will be as effective and, ideally, as cheap to produce as praziquantel. The approaches to this problem currently in progress can be grouped into several categories: the use of antimalarials (Shu-Hua et al. 2006, Manneck et al. 2011), the benzodiazepines (Baard et al. 1979) (first investigated before the advent of praziquantel and now being revisited), antioxidant enzyme inhibitors (Sayed et al. 2008) and finally the use of anti-cancer drugs and strategies. Ethnopharmacological approaches, based on plants used by traditional healers in endemic areas, remain relatively unexplored (Togola et al. 2008). All these strategies have advantages and disadvantages. Artemisinins are front-line drugs for the treatment of malaria and artemisinin-based combination therapy is now the recommended treatment strategy (Eastman & Fidock 2009). They are also effective against schistosomes and, in contrast to praziquantel, kill the larval stages in vivo (Utzinger et al. 2002). However, their use against schistosomiasis is discouraged since there is a risk of promoting resistance of Plasmodium falciparum in endemic areas where schistosomiasis and malaria overlap (Keiser & Utzinger 2007). Interest in the benzodiazepines was originally diminished due to their side-effects (impairments to psychomotor functions, sedation and ataxia) (O'Boyle et al. 1985). However, the fact that these could be abolished by treatment with flumazenil meant that meclonazepam could be reconsidered as a lead compound and is now actively investigated (Thibaut et al. 2009). "Achilles heel" approaches based on targeting vital enzyme activities of the parasite are relatively new. The characterization of oxadiazoles as powerful inhibitors of Schistosoma mansoni thioredoxin glutathione reductase with activity against the parasite in vivo opened up a new class of compounds for investigation (Sayed et al. 2008). The search for potential drug targets has been facilitated and stimulated by the availability of the genome sequence for two schistosomes (Berriman et al. 2009, The Schistosoma japonicum Genome Sequencing and Functional Analysis Consortium 2009). Three strategies based on the genomic data are possible. First, the search for schistosome-specific molecules in vital pathways would in theory permit the development of drugs with no adverse effect on the host. Second, the identification of orthologues of proteins that have previously been shown to be targets of anthelminthics, such as ion channels (for instance the voltage-gated Ca2+ channel that is a putative target of praziquantel) (Jeziorski & Greenberg 2006) and neurotransmitter receptors, such as the acetylcholine receptor targeted by levamisole (Martin & Robertson 2007 for review) might allow the use of the existing drugs as lead compounds. Finally we can seek to identify orthologues of proteins already targeted in other pathologies. By this latter approach, which has been termed "piggy-backing" (Dissous & Grevelding 2011), anti-cancer drugs, such as imatinib, that target protein kinases, can be used as lead compounds for treating schistosomes since they express orthologues of the target proteins. Indeed, imatinib has been shown to kill adult schistosomes in vitro and, prior to parasite death, the drug affects morphology and causes separation of worm couples (Beckmann & Grevelding 2010). Our approach is similar, since we propose to consider schistosomes themselves as cancerous growths: they have an intense metabolic activity and a high rate of cell division (for the production of eggs by female worms) that is outside the control of the host. We are therefore concentrating on classes of enzymes that are shared with the human host, but can be specifically targeted in cancer cells; inhibition of these enzymes arrests growth or induces apoptosis in cancer cells, but not in normal cells. These are enzymes that modify histones and thereby alter the state of the chromatin, inducing activation or repression of gene transcription. Histone modifying enzymes (HME) as drug targets - HME include histone deacetylases (HDAC) that have been intensively studied as drug targets, but other classes of enzyme, including histone acetyltransferases (HAT), histone methyltransferases and histone demethylases are increasingly investigated. HME are central actors in the regulation of the epigenetic modification of chromatin and aberrant epigenetic states often associated with cancer led to interest in HME as targets for therapy. HDACs deacetylate acetylated lysine residues in a variety of proteins, including histones, but also transcription factors and cofactors, as well as non-nuclear proteins such as tubulin. There has been a considerable effort to develop HDAC inhibitors (HDACi); a number are in clinical trials and two are currently approved for use by the US Food and Drug Administration. Suberoylanilide hydroxamic acid (SAHA, vorinostat) has been approved for use in adults with cutaneous T-cell lymphoma (CTCL) (Duvic et al. 2007) and extensively tested against other types of cancer either alone or in combination with other agents (Wagner et al. 2010 for review). A second HDACi, depsipeptide (FK228, Romidepsin) was also approved initially for use in CTCL. Broadly, HDACi induce cell death in cancer cells via apoptosis (see below), but they can also act on the cell cycle, on tumour angiogenesis or via the regulation of host cell responses (Bolden et al. 2006 for review). HDACi have also stimulated interest as anti-parasitic drugs and have been tested against P. falciparum (Andrews et al. 2000, 2008), Toxoplasma gondii (Strobl et al. 2007) and the major kinetoplastid parasites (Mai et al. 2004, Horn 2008). In the case of P. falciparum, it was possible to develop HDACi that are significantly more toxic to the parasite than toward human cells (Andrews et al. 2008, Wheatley et al. 2010). We have shown (Dubois et al. 2009) that HDACi such as trichostatin A (TSA) and valproic acid (VPA) cause the death of S. mansoni larvae and adult worms in vitro and that this is probably via the induction of apoptosis in the parasites (see below). Schistosome HDACs, as well as other HME, are therefore promising targets for the development of new drugs. The nucleosome, the basic unit of chromatin, is formed by an octamer of four histones around which 147 bp of DNA are wound. The histone proteins (H2A, H2B, H3 and H4) consist of a globular domain and an unstructured tail domain. These tails can undergo a variety of post-translational modifications (acetylation, methylation, phosphorylation, ubiquitinylation, sumoylation etc.) that affect the overall chromatin structure and the transcription of genes and together make up the "histone code" (Jeunwein & Allis 2001). Two of these modifications seem to be particularly important in affecting the higher order structure of the chromatin fibre and in the consequent regulation of transcription: acetylation of lysine residues and the methylation of lysines or arginines of the tails of histones [Fig. 1]. In this review we will focus on the role of the acetylation/deacetylation balance and inhibitors of the enzymes involved as drug candidates. The acetylation or deacetylation of conserved lysine residues in the amino-terminal tails of histones represents a crucial element in the chromatin-based mechanisms of transcriptional regulation and the histone code. Broadly, acetylation via HAT annuls the positive charge of the lysine and reduces chromatin compaction, favouring transcription, whereas deacetylation, via HDAC has the opposite effect [Fig. 2]. However, acetylation also provides binding sites for bromodomain-containing transcriptional effectors (Jeunwein & Allis 2001). It is also an over-simplification to suggest that HDAC are always associated with transcriptional repression (Zupkovitz et al. 2006). The enzymes involved in these processes also have roles in the deacetylation of other proteins, actors in transcriptional complexes or in some cases, cytosolic proteins such as tubulin. A large number of transcriptional coactivators are now recognized to possess HAT activity and it is possible to distinguish at least six families of HAT proteins (Sterner & Berger 2000): the GNAT superfamily (PCAF/GCN5), the MYST family, the CBP/p300 family, the nuclear receptor coactivator (SRC/ACTR/TIF) family, the TAFII 250 family and the GNAT related family (TFIIIC). An orthologue of the HAT, GCN5 from S. mansoni was shown to possess conserved catalytic properties (de Moraes Maciel et al. 2004) and to specifically acetylate lysine 14 of histone H3 (de Moraes Maciel et al. 2008). In addition, two orthologues of mammalian CBP/p300 that also had conserved HAT activities have been characterized (Bertin et al. 2006). HDAC in eukaryotes have traditionally been divided into three classes, with classes I and II comprising enzymes that share similar catalytic domains and a Zn2+-dependent catalytic mechanism. More recently, a separate class (IV), comprising only HDAC 11 in mammals, was described (Gregoretti et al. 2004). Class III comprises the enzymes related to yeast Sir2 that are NAD+-dependent and phylogenetically unrelated to classes I and II. In common with Gregoretti et al. (2004), these will be referred to hereafter as sirtuins. HDACs and sirtuins are members of ancient enzyme families that are present in achaebacteria, eubacteria, protists, plants, fungi and animals. HDAC class I is represented in mammals by HDACs 1, 2, 3 and 8 and are generally smaller than the class II HDACs (4-10) with only relatively short N and C-terminal domains surrounding the catalytic domain. Class II HDACs have generally larger N or C-terminal domains and HDAC6 has two catalytic domains. Indeed HDAC6 is grouped with HDAC10, which has a second, truncated, catalytic domain in a sub-class IIb. HDAC 1 forms homo or heterodimers (with HDACs 2 or 3) and HDAC 3 also interacts with class II HDACs (Gregoretti et al. 2004). In a preliminary study, we cloned and characterized three class I HDACs present in the S. mansoni genome, orthologues of mammalian HDACs 1, 3 and 8, and confirmed their identities by phylogenetic analysis (Oger et al. 2008). The identification of an orthologue of HDAC8 in an invertebrate was unexpected as it had been suggested that this was a vertebrate-specific enzyme (Gregoretti et al. 2004). All three have conserved catalytic domains, but divergent C-terminal domains. Insertions in the SmHDAC8 catalytic domain indicate that it may exhibit specific enzymatic properties compared to the mammalian counterpart. Structure-guided optimization strategies could take into account these differences. SmHDAC 1, 3 and 8 mRNAs are expressed at all life-cycle stages, SmHDAC3 being expressed at a lower level than SmHDAC1 and 8. SmHDAC1 was shown to repress NF-ΚB and Gal4-dependent transcriptional activity in a mammalian cell line. This activity was dependent on the catalytic activity of SmHDAC1 since transcription was partially restored by treatment with the HDAC inhibitor TSA and a catalytic site mutant form of SmHDAC1 failed to repress transcription (Oger et al. 2008). A preliminary in silico study also allowed the identification of three class II HDACs encoded in the S. mansoni genome, orthologues of HDACs 4, 5 and 6. The full-length coding sequences and splice variants of each of these HDACs has been verified using rapid amplification of cDNA ends-polymerase chain reaction (RACE-PCR) (unpublished observations) and the characterization of their expression profiles during the S. mansoni life-cycle is under investigation. Sirtuins can be separated into four main classes (with a 5th class present in Gram-positive bacteria) (Frye 2000) and the human sirtuins (7 in all) comprise members of all four groups. Sirtuins (Sirt) 3, 4 and 5 are localized in the mitochondria, Sirts 6 and 7 are exclusively nuclear, Sirt 1 has a dual nuclear/cytosolic localisation and Sirt 2 is cytosolic (Michishita et al. 2005). In addition to their lysine deacetylase activity, sirtuins metabolize NAD and can act as mono-ADP-ribosyltransferases. Indeed Sirt4 is a mitochondrial enzyme that ADP-ribosylates and down-regulates glutamine dehydrogenase (Haigis et al. 2006). Five sirtuins are encoded in the S. mansoni genome, orthologues of human sirtuins 1, 2, 5, 6 and 7 (unpublished observations). Their predicted localizations (PSORTII) (Nakai & Horton 1999) are similar to those of the human orthologues. HAT and HDACi - Most efforts to develop drugs based on HMEs have concerned the HDACs, but both HAT and HDACi have either anti-proliferative effects or cause apoptosis in tumour cell lines. Derivatives of γ-butyrolactone were designed as GCN5 inhibitors and high throughput screening led to the identification of isothiazolones as inhibitors of P300/CBP-associated factor (PCAF) and p300 (Stimson et al. 2005). More recently, chemicals derived from medicinal plants such as anacardic acid, garcinol and curcumin were found to be potent inhibitors of p300 and PCAF and the two latter chemicals induced histone hypoacetylation in cancer cells, leading to apoptosis (Balasubramanyam et al. 2004). Inhibitors of class I and II HDACs can be classed in four families according to their structure: short-chain fatty acids (butyric acid derivatives including VPA), hydroxamic acid derivatives, including TSA and SAHA, that were first detected for their capacity to induce apoptosis in screens on transformed cells before being shown to be HDACi (Richon et al. 1998), benzamides and cyclic tetrapeptides. Representatives of each of these families are currently in clinical trials against diverse cancers and half maximal inhibitory concentration values for these inhibitors are generally in the µM or nM range (Wagner et al. 2010). In cancer therapy, HDACi have proved to have potent activities at concentrations that are minimally toxic to the host, although they do have side-effects (Bolden et al. 2006, Wagner et al. 2010 for reviews). HDACi selectively kill cancer cells in vitro at concentrations 10-fold lower than for normal cells. The effects of HDACi are cell-type dependent and the molecular pathways engaged to mediate their effects are not fully elucidated. However, they are capable of inducing apoptosis via several pathways, including death receptors, the mitochondrial pathway, selective activation of BH3-only proteins or via the regulation of the production of reactive oxygen species. Many HDACi can induce cell-cycle arrest at G1/S and they inhibit cancer vascularisation, possibly via the downregulation of the expression of the chemokine receptor CXCR4. Finally, HDACi can enhance antitumour immunity, either by rendering malignant cells more visible to the immune system or by altering immune cell activity and/or cytokine production (Bolden et al. 2006). Less is known about the consequences of inhibiting sirtuins; only a limited number of inhibitors have been described and some do not inhibit human subtypes. The first synthetic inhibitor described was sirtinol, but precipitation of the enzyme by the inhibitor may contribute to in vitro inhibition. Indoles active down to 0.1 µM have been described and the anticancer activity of one of these, cambinol, was shown to be effective in an animal model (Heltweg et al. 2006). Splitomicin derivatives have been characterized that inhibit recombinant Sirt2 and have antiproliferative properties and promote tubulin hyperacetylation in a breast cancer cell line (Neugebauer et al. 2008). In addition, thiobarbiturates have been recently discovered to be a novel class of sirtuin inhibitor (Uciechowska et al. 2008). HDACi as drugs against parasites - Parasites can be likened to tumours in that they undergo intense metabolic activity that is outside the control of the host. Schistosomes do not proliferate within the human host, but have in common this intense metabolic activity and a high level of proliferation of the vitelline cells involved in the massive production of eggs. Moreover, parasites, like tumours, render themselves invisible to the immune system, either by means of genetic variation as in the case of malaria parasites (Templeton 2009), or like schistosomes, by adsorbing host determinants onto their surface (Sher et al. 1985). HDACi and sirtuin inhibitors have therefore been tested for their activity against a variety of parasites. The use of HDACi against malaria parasites started with the demonstration of the activity of apicidin (a cyclic tetrapeptide HDACi) in inhibiting growth of P. falciparum in vitro. Subsequent work showed that TSA was also active in vitro and that suberic acid bisdimethylamide had a cytostatic effect on the murine malaria parasite Plasmodium berghei in vivo (Andrews et al. 2000). Subsequently, compounds derived from L-cysteine or 2-aminosuberic acid were designed to inhibit P. falciparum HDAC-1 based on homology modelling with human class I and class II HDAC enzymes (Andrews et al. 2008). These compounds showed a high antiproliferative activity in the nM range and some were much more toxic toward the parasites than toward mammalian cells. More recently, Wheatley et al. (2010) described a series of inhibitors with cinnamic acid derivatives or non-steroidal anti-inflammatory drugs as HDAC-binding motifs. These were found to be up to 10 times more toxic toward P. falciparum than a normal human cell line. This work underlines the possibility of designing inhibitors with increased specificity toward HDACs of parasites via molecular modelling. Similarly, a variety of hydroxamic acid class HDACi, including TSA and SAHA at nM concentrations, were capable of inhibiting proliferation of the apicomplexan parasite T. gondii in vitro and completely protected monolayers of HS68 cells against infection (Strobl et al. 2007). Moreover, in the case of the kinetoplastid parasite Trypanosoma brucei, an apicidin analogue has been shown to have potent and selective antiproliferative effects (Murray et al. 2001). The inhibition of sirtuins has been less investigated for its therapeutic potential in protozoan parasites. Although P. falciparum Sir2 was inhibited by nicotinamide, a natural product of the sirtuin reaction, it was not sensitive to sirtinol or splitomicin (Merrick & Duraisingh 2007). However, recent studies (Chakrabarty et al. 2008, Prusty et al. 2008) have shown that both nicotinamide and the synthetic inhibitor surfactin inhibit PfSir2 activity and are potent inhibitors of intra-erythrocytic growth of the parasite. Chakrabarty et al. (2009) have also developed lysine-based tripeptide analogues as PfSir2 inhibitors, one of which also markedly inhibited parasite intra-erythrocytic growth. Moreover, sirtinol was found to inhibit the in vitro growth of Leishmania infantum via the induction of apoptosis (Vergnes et al. 2005). Structural studies of Leishmania Sir2 (Kadam et al. 2008) have recently (Tavares et al. 2010) led to the characterization of bisnaphthalimidopropyl derivatives as inhibitors and potential drugs. The use of HAT inhibitors has been more limited, but anacardic acid has been shown to have parasiticidal effects on P. falciparum that are associated with changes in global gene expression (Cui et al. 2008). An in silico screening approach using a modelled P. falciparum HAT suggested the use of curcumins and diarylheptanoids as potential lead compounds (Singh & Misra 2009). Effects of HDACi on schistosomes - The inhibitors of class I and II HDACs, VPA SAHA and TSA both inhibited global HDAC activity in schistosomes (Dubois et al. 2009). When used to investigate the effects of HDAC inhibition on schistosomula and adult worms maintained in in vitro culture, TSA and VPA both caused dose dependent mortality of schistosomula and adults, with TSA showing the most marked effect (range from 1-10 µM). TSA treatment of schistosomula led to an increase in caspase 3/7 mRNA expression and enzyme activity that was mirrored by TUNEL labelling of larvae, showing that apoptosis was induced (Dubois et al. 2009). Both TSA and VPA were shown to cause an increase in general levels of protein acetylation in schistosomes; more particularly of histone 4, whereas histone 3 acetylation was much less affected. This general increase in acetylation was shown to be accompanied by an increase in the expression of the genes encoding orthologues of caspases 3 and 7, but not of the SmHDAC1 gene. The caspase 3 gene has previously been shown to be upregulated in cell lines after treatment with SAHA (Tan et al. 2006), human HDAC1 is upregulated by TSA (Zupkovitz et al. 2006) and the absence of upregulation of SmHDAC1 in schistosomula was surprising. However, ChIP analysis of H4 acetylation in the promoters of these genes showed that the SmHDAC1 promoter is already maximally acetylated in untreated parasites, whereas the caspase 3 and 7 gene promoters showed significantly increased acetylation after TSA treatment. Together, these results showed that, as with tumour cell lines, HDACi induced hyperacetylation of histones, deregulated gene expression and induced apoptosis in schistosomes. In order to determine whether schistosome sirtuins might also be considered drug targets, we have recently treated schistosomula in culture in the presence of sirtinol. This compound proved a potent inducer of parasite death and this correlated with a positive TUNEL assay [Fig. 3]. The challenge - the Schistosome Epigenetics: Targets, Regulation, New Drugs (SEtTReND) project (settrend.cebio.org/) - Schistosome HDACs, sirtuins and by extension, other HMEs are clearly potential drug targets. However, there are several challenges to overcome in order to develop drugs based on inhibitors of these enzymes. The inhibitors have to be made as specific as possible for the schistosome enzymes in order to minimize side-effects that can be caused either by reactivity with the human enzymes or by off-target effects. They have to be bio-available and to be administered by the oral route to render mass-treatment possible. And they have to be cheap to produce. Rather than approach this problem piece-meal by looking at possible targets and drugs in isolation, we have decided to adopt a holistic approach to drug development based on the HMEs. A project, called SEtTReND, was developed with nine partner teams based in Europe and Brazil that has received financial support from the European Commission (FP7-Health). The different approaches to be undertaken during the project are summarized in photo/oc11165f4.jpg">[Fig . 4]. They include: (i) the identification of all the enzymes involved in histone acetylation/deacetylation and methylation/demethylation by mining the schistosome genome database and by validation of the predicted sequences using RACE-PCR and molecular cloning; (ii) phenotypic screening using established inhibitors of the different HME subclasses on schistosomes in culture, combined with the TUNEL assay, in order to refine the choice of enzyme classes as targets; (iii) validation of target HMEs by RNA interference and determination of an apoptotic phenotype. This will be combined with the determination of gene expression signatures of interfered schistosomes to generate a profile corresponding to specific inhibition of a target. This can be compared with signatures generated after drug treatment to determine drug specificity and potential off-target effects; (iv) the production of pure recombinant enzymes for testing inhibitors and for 3D structural determination (X-ray crystallography); (v) high-throughput screening on recombinant enzymes with compound libraries combined with fragment-based screening; (vi) a parallel rational drug-design approach based on modelling the catalytic pocket of the enzymes and in silico screening, followed by optimization; (vii) lead compounds generated by either or both of these two last approaches will be tested on schistosomes in culture and in infected mice. Successful leads will then go forward for optimisation (specificity, bio-availability). The issue of specificity is clearly crucial when dealing, as here, with enzymes that have functions conserved throughout evolution. All the more so when considering that schistosomes are metazoans and not as phylogenetically distant from humans as the protozoan parasites, such as P. falciparum, for which apparently specific HDACi have been developed. Among the S. mansoni class I HDACs that we characterized (Oger et al. 2008) SmHDAC1 and 3 have highly conserved catalytic domains and it would be difficult to develop specific inhibitors for these enzymes. SmHDAC8, however, is phylogenetically distant from its orthologues (mammalian or insect) and has several insertions in the catalytic domain. Moreover, the predicted subcellular localization of SmHDAC8 is cytosolic, in contrast to the nuclear localization of the human orthologue. This suggests that the schistosome enzyme may have a different role and different substrates. Similarly, the sirtuins SmSirt1 and 2 show notable differences in their catalytic domains compared to mammalian orthologues (not shown) and it is therefore probable that other schistosome HMEs will show sufficient differences in order to render possible the development of specific inhibitors. Therefore, the combination of high-throughput and rational approaches to drug discovery, followed by optimisation of the lead compounds, should allow us to validate this strategy as a paradigm for drug development against schistosomes. ACKNOWLEDGEMENTS To the SEtTReND consortium, for help and support. REFERENCES
Received 26 April
2011 Copyright 2011 - Memorias do Instituto Oswaldo Cruz The following images related to this document are available:Photo images[oc11165f4.jpg] [oc11165f1.jpg] [oc11165f2.jpg] [oc11165f3.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
![[Fig . 4]](/showimage?oc/photo/oc11165f4.jpg){kind=link}