 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
RESEARCH NOTE Simplified method for preservation and polymerase chain reaction-amplification of Trypanosoma cruzi DNA in human blood Patricia L Dorn+, Simona Selgean, Monique Guillot Department of Biological Sciences, Loyola University, 6363 St. Charles
Avenue, New Orleans, LA 70118 U.S.A. Received 17 June 1996
Code Number:OC97050
Sizes of Files:
Text: 12.6K
Graphics: Photographs (jpg) - 18.3K
Key words: Trypanosoma cruzi - Chagas' disease - polymerase chain reaction - DNA preservation - human blood The polymerase chain reaction (PCR) looks to be a promising method for diagnosis of Trypanosoma cruzi infection and has advantages over current methods (serology, direct microscopic examination, hemoculture and xenodiagnosis) in that PCR can distinguish between infection with other hemoflagellates, does not depend on the immunocompetence of the patient, is sensitive enough for use in chronic patients, and can distinguish a current infection from a previous one or a newborn infection from a maternal one (HA Avila et al. 1993 J Clin Microbiol 31: 2421-2426, C Britto et al. 1995 Parasitol 110: 241-247). Preservation of T. cruzi DNA in the blood sample, which is often drawn in a remote area and processed for PCR several days later, is an important aspect of an assay useful in endemic areas. Blood sample preservation has previously been accomplished by collecting the blood in a guanidine/EDTA solution which preserves the parasite DNA at 37 C for at least one month (HA Avila et al. 1991 Mol Biochem Parasitol 48: 211-222). Following collection, the samples are boiled to disperse the template throughout the lysate, thus increasing the probability that a sample taken for PCR will contain at least one template (C Britto et al. 1993 Mem Inst Oswaldo Cruz 88: 171-172). Since guanidine is a strong protein denaturant, to allow the sample to be amplified by the PCR, all the guanidine must first be removed which is accomplished by phenol extraction. This protocol, although effective, necessitates working with the hazardous chemicals guanidine and phenol and extensive template handling which increases the risk of cross-contamination. In addition, phenol extraction leads to variable recovery of sample (unpublished data), such that following a single phenol extraction, not all the samples containing parasites can be amplified (P Wincker et al. 1994 Am J Trop Med Hyg 51: 771-777). We show here that DNA in blood may be effectively preserved by high levels of EDTA up to two weeks at 37 C and is a suitable template for amplification for at least three months. T. cruzi-specific PCR primers have been developed for nuclear repetitive RNA or DNA (DR Moser et al. 1989 J Clin Microbiol 27: 1477-1482, JM Requena et al. 1992 Mol Biochem Parasitol 51: 271-280, RP Souto & B Zingales 1993 Mol Biochem Parasitol 62: 45-52, N Gonzales et al. 1994 J Clin Microbiol 32: 153-158) or kinetoplast DNA (kDNA), part of the unusual mitochondrial DNA found in trypanosomes (NR Sturm et al. 1989 Mol Biochem Parasitol 33: 205-214, F Veas et al. 1991 Cell Mol Biol 37: 73-84). kDNA minicircles are an ideal target for PCR amplification since they are present in 10-20,000 copies per parasite. The PCR assay based on kDNA is highly specific for T. cruzi: Leishmania and T. brucei are not amplified and T. rangeli, although amplified, shows a different-sized band by gel electrophoresis. PCR primers, which are complementary to the ends of highly conserved regions of the kDNA minicircle, will potentially amplify all T. cruzi isolates. An added advantage is that the region amplified includes the hypervariable region of the kDNA which may be used for strain typing of T. cruzi isolates using strain-specific probes from this region or schizodeme analysis (Veas loc. cit., Sturm loc.cit.). We show that using modified kDNA-specific primers in an optimized PCR reaction, approximately 100 T. cruzi minicircles (equivalent to approximately 5/1000th of a parasite) may be detected in blood preserved in EDTA.
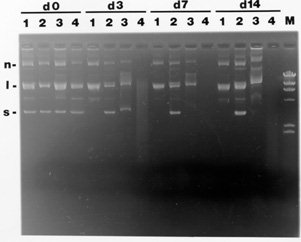
To test the effectiveness of different preservatives for maintaining the quality of DNA in blood, plasmid DNA (a pUC-based plasmid) was added to blood containing the preservatives to be tested and the quality of the DNA was assessed over time. To mimic tropical field conditions, mixtures were kept at 37 C for the times indicated, then processed by phenol:chloroform:isoamyl alcohol (25:24:1) extraction, ethanol precipitation and visualized by ethidium bromide-stained agarose gel electrophoresis. The results of storage of plasmid DNA in blood at 37 C with different concentrations of EDTA, sodium citrate, or guanidine/EDTA is shown in Fig. 1. At day 0, all three forms of plasmid DNA: supercoiled circular (s), linear (l) and nicked circular (n) are evident in all of the mixtures of plasmid DNA, blood, and preservative. By day 3, the 0.25 M EDTA and guanidine effectively preserve the plasmid DNA as shown by maintenance of the supercoiled circular form (d3, lanes 2 and 3, respectively) whereas the plasmid DNA in 0.1M EDTA is no longer in supercoiled form but only in linear and nicked circular forms (d3, lane 1) and the plasmid DNA in sodium citrate has been completely degraded (d3, lane 4). Following one and two weeks, the higher level of EDTA appears to be the most effective treatment for retaining the supercoiled circular DNA (d7 and d14, lane 2), even more effective than the current method of guanidine/EDTA (d7 and d14, compare lanes 2 and 3). Later work showed that 0.2M EDTA was sufficient for preservation so this level was used in all subsequent studies. Once we had shown that DNA is effectively preserved in EDTA, we attempted to amplify the blood/EDTA mixture directly by compensating for the high levels of EDTA with additional equimolar amounts of MgCl2 or ZnSO4 in the PCR reaction. However, we rarely saw amplification and so pursued a method of removing the EDTA. We removed the EDTA with either a simple, approximately 20 min DNA column isolation (QIAamp, performed according to manufacturer's instructions, QIAGEN, CA, U.S.A.) or buffer exchange on Microcon-30 columns (performed according to manufacturer's instructions, Amicon, Beverly, MA, U.S.A.). For both methods, 2ul of parasites (epimastiogotes, strain QRO, a gift of Dr Paz Maria Salazar, UNAM, Mexico), were added to 1 ml of a blood/EDTA mixture (0.5 ml 0.4M EDTA, 0.5 ml of blood, for a final EDTA concentration of 0.2M). 200ul of this solution was boiled for 15 min, centrifuged, the supernatant (approximately 100ul) was processed by QIAamp or Microcon columns, and 5ul removed for the PCR. To test the level of sensitivity of the assay, blood with parasites and EDTA was boiled and combined with previously boiled blood/EDTA to give the correct number of parasites in each sample. For the PCR, we modified the primers of Veas et al. (loc. cit.), to better match the published T. cruzi minicircle sequences, to preserve the Sau96I site, and to incorporate an HphI site [primers TC1:
5'-TTGAACGGCCCTCCCAAAAC-3' and
-----
TC2: 5'-GATTGGGGTTGGTGAAATATA-3';
-----
bolded nucleotides indicated changes from Veas et al. (loc. cit.),
underlined nucleotides indicate the restriction sites]. The restriction
sites were used later for isolation of the hypervariable region. Our
optimized PCR assay is as follows: 5ml of DNA template, 10mM Tris-HCl, pH
9.0, 0.1% Triton X-100, 75mM KCl, 3.5mM MgCl2, 200mM each dNTP, 20 pmoles
each primer and 2 U Tth DNA polymerase (Promega, chosen because it
is less inhibited by blood than is Taq polymerase). An initial
denaturation step of 94 C for 3 min was followed by 35 cycles of 94 C, 55 C
and 72 C, each for 1 min, followed by a 10 min extension at 72 C (MJ
Research Programmable Thermal Controller, Watertown, MA, U.S.A.). 20% of
the PCR reactions were electrophoresed on a 1.8% agarose gel and visualized
by ethidium bromide staining.
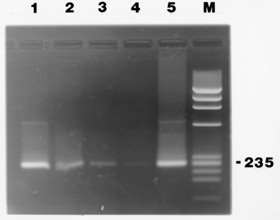
This PCR assay, when used with purified kDNA in water, detects 10^-4 fg of kDNA and can be used to amplify strains from diverse geographic regions including: Mexico, Bolivia, Columbia and the U.S.A. (data not shown). Results of the sensitivity study in blood are shown in Fig. 2. The expected 235 bp band is present in all of the lanes (50 to 0.005 parasites, lanes 1-4). Thus, we are able to amplify as little as ~100 minicircles in blood (equivalent to approximately 5/1000th of a parasite) preserved in 0.2M EDTA following boiling and QIAamp processing (identical results were obtained with the Microcon columns). To test how long an infected blood sample could be preserved in EDTA and still be amplified by PCR, we combined blood from a healthy individual with EDTA (at 0.2 M) and T. cruzi parasites at 10^4 parasites/ml. The mixture was incubated at 37 C, 200ul aliquots were removed weekly for four weeks and then monthly out to three months, and processed for PCR using the QIAamp method as described above. A T. cruzi-specific band (235-bp) was evident in all samples tested including the latest (3 months) which is shown in Fig. 2, lane 5. Recent results have shown that DNA is also amplified when the blood sample is processed using StrataClean resin (Stratagene, CA, U.S.A.), which will provide a less expensive alternative to the QIAamp method. Results shown here demonstrate that parasite DNA may be preserved in 0.2M EDTA out to three months and that less than one parasite is successfully amplified by the PCR using modified primers and the template preparation described here. This was work supported by the American Heart Association-Louisiana Affiliate, the Howard-Hughes/Hope College Fund, Loyola Student Government Association-Richard Frank Grant, and the Loyola University Faculty Development Grant. Copyright 1997 Fundacao Oswaldo Cruz The following images related to this document are available:Photo images[oc97050b.jpg] [oc97050a.jpg] |
| |||||||||

{kind=link}
{kind=link}