 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Separation and Mapping of Chromosomes of Parasitic Protozoa Rosaura Hernandez-Rivas, Artur Scherf^+ Unite de Parasitologie Experimentale, CNRS URA 1960, Institut Pasteur,
25 Rue du Docteur Roux, 75724 Paris Cedex 15, France
This work has been supported by a grant from Groupement de Recherches et d'Etudes sur les Genomes (GIP-GREG). R H-R was financed by fellowships from `Pasteur-Weizmann' and `Fondation pour la Recherche Medicale'. Received 20 August 1997; Accepted 10 September 1997
Code Number:OC97153
Size of files:
Text: 18.2K
Graphics: Table (jpg) - 16.2K
Line drawings and photographs (jpg) - 22.6K
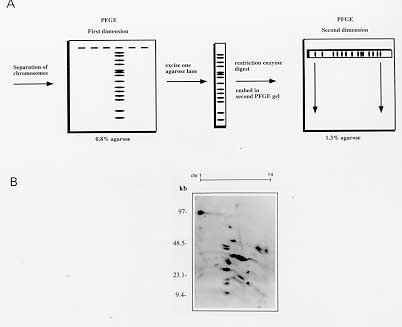
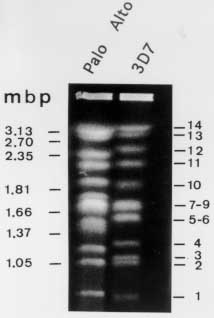
Many protozoan parasites represent an important group of human pathogens. Pulsed Field Gradient Gel Electrophoresis (PFGE) analysis has been an important tool for fundamental genetic studies of parasites like Trypanosoma, Leishmania, Giardia or the human malaria parasite Plasmodium falciparum. We present PFGE conditions allowing a high resolution separation of chromosomes ranging from 500 to 4000 kb within a two day electrophoresis run. In addition, we present conditions for separating large chromosomes (2000-6000 kb) within 36 hr. We demontrate that the application of two dimentional PFGE (2D-PFGE) technique to parasite karyotypes is a very useful method for the analysis of dispersed gene families and comparative studies of the intrachomosomal genome organization. Key words: Pulsed Field Gradient Gel Electrophoresis - two dimensional PFGE - protozoan karyotypes - chromosome mapping - dispersed gene families In 1984, Schwartz and Cantor introduced a new technique in which periodic changing of the direction of the electric field at fixed time intervals allowed the separation of large DNA molecules (>50 kb), called Pulsed Field Gradient Gel Electrophoresis (PFGE). Since then, various improvements of the PFGE technique have been made allowing for the separation of DNA molecules up to 13 Mb in agarose gels (for review Lognonne 1993). Protozoa have relatively small genomes ranging generally from several kilo base pairs to several mega base pairs. Classical genetic studies have been hampered by the absence of chromosome condensation during metaphase. Thus, PFGE analysis has been a very important tool for fundamental genetic studies of protozoan parasites such as Trypanosoma (Van der Ploeg et al. 1984), Leishmania (Giannini et al. 1986), Giardia (Adam et al. 1988) or the human malaria parasite Plasmodium falciparum (for review see Foote & Kemp 1989). PFGE applications are successful in resolving karyotypes, demonstrating chromosome polymorphism and mapping of chromosomes of many pathogens. Most protozoan parasites have chromosome sizes that can readily separated by PFGE. However, due to the complexity of many parasite karyotypes, the separation of chromosomes by PFGE of many of these pathogenic unicellular parasites can take prolonged electrophoresis time. For example, the time to separate the 14 chromosomes of P. falciparum has been reported to be one week (Dolan et al. 1993). In this paper, we present protocols for the preparation, separation and digestion of agarose embedded intact high molecular weight DNA. We describe also recently improved PFGE conditions allowing high resolution separation of chromosomes of most parasitic protozoa within a two day electrophoresis run (Hinterberg & Scherf 1994). MATERIALS AND METHODS SeaKem GTG agarose (FMC BioProducts) for agarose gels concentrations >0.6% and chromosomal grade agarose (Bio-Rad) for agarose gels <0.6%. Low-melting point agarose (FMC BioProducts) for embedding parasites in agarose blocks and preparative isolation of chromosomes. Molecular weight size standards: 5 kb ladder (4.9-120 kb), lambda ladder (0.05-1 Mb), Saccharomyces cerevisiae (0.2-2.2 Mb) and Hansenular wingei (1-3.1 Mb) (Bio-Rad). Erythrocyte lysis buffer: 0.15% saponin (Sigma) in phophate buffered saline (PBS). Cell lysis buffer (CLB): 0.5M EDTA pH 8.0, 0.01M Tris-HCl, 1% sodium lauryl sarcosinate (Sigma). Store at room temperature. Before cell lysis add 2 mg/ml of Proteinase K from a stock solution. Ethidium bromide: 10 mg/ml in water. Nylon membrane: Hybond N^+ (Amersham) or BIOTRANS^(+) (ICN). These membranes allow the rapid alkaline transfer of DNA and the physical characteristics make them specially useful for multiple hybridization cycles (usually 5-10 cycles). Alkaline transfer buffer: 0.4 M NaOH. Prehybridization and hybridization buffer: 0.5M NaPO4, pH 7.2, 7%SDS, 1% BSA. 20xSSC: 3M sodium chloride, 0.3M sodium citrate. Saturated sec-butanol: mix sec-butanol with 1M NaCl in TE (4:1 v/v). Add 9 vol saturated sec-butanol and 1 vol of the aqueous phase to the gel blocks. This is important to avoid gel shrinking! 10xTBE buffer: 108 g Tris base, 54 g boric acid, 8.35 g disodium EDTA per liter (pH approx. 8.5). TE buffer: 10 mM Tris-HCl, 1 mM EDTA pH 8.0. Random prime labeling kit (Boehringer Mannheim or Amersham). alpha-^32P-dATP, >3000 Ci/mmol. Preparation of DNA for PFGE - The ability to preserve the intact size of large DNA molecules is critical for the success of PFGE. Very high molecular weight DNA samples are impossible to prepare by conventional solution methods because they are extremely sensitive to shear damage. To overcome this problem, intact cells are embedded in low melting agarose blocks (Schwartz & Cantor 1984). Cells are lysed and proteins are removed by Proteinase K treatment. This procedure yields DNA that is both intact and susceptible to restriction enzyme digestion. The agarose block can be loaded directly into the well of a pulsed-field gel. The host cell of the intracellular bloodstage form of P. falciparum needs to be lysed by saponin before embedding the parasite cells into agarose blocks. The first step of the protocol concerns only the preparation of chromosomes of plasmodial bloodstage parasites. If you work with free living parasites start with step 2. 1. Estimate the volume of the erythrocyte pellet and add 1.5 vol of 0.15% saponin in PBS at room temperature. Resuspend the pellet and incubate for 3-5 min. The liquid will clarify as the red blood cells lyse. Add 5 vol of cold PBS and centrifuge at 5000 rpm for 10 min. Resuspend the dark pellet which contains the parasite material in 2 ml PBS. 2. Pellet the cells by centrifugation at 3000 rpm for 10 min and resuspend at a concentration of approx. 5x10^8 parasites/ml PBS. 3. Equilibrate the cell suspension at 37 C and add an equal volume of 1.6% melted low-melting point agarose (37 C). Mix gently and dispense the cell suspension immediately into plastic gel molds (2x5x10mm, 100 ml vol) (Pharmacia) and allow to solidify at 4 C for about 20 min. 4. Place up to 20 solidified blocks in a volume of 10 ml containing cell-lysis buffer (CLB) and incubate for 24 hr at 42 C. Replace the CLB with an equal volume of fresh CLB and incubate for additional 24 hr. 5. The blocks can be stored indefinitely in TE buffer at 4 C. Electrophoresis conditions - Optimal separation of the different chromosome size classes usually requires the use of a range of different PFGE conditions. Table shows the running conditions of large DNA fragments of various sizes using the CHEF system. Several parameters that influence the DNA mobility, such as the temperature, the voltage, pulse time and agarose type have been modified to enhance the mobility of the chromosomes. The use of lower agarose percentages, higher output voltage in combination with one or two phase ramping switch time protocols reduced significantly the time of separation of chromosomes ranging from about 600 to 3500 kb. A summary of run conditions for various size ranges are given in Table. The presented PFGE methods have been used in studies of P. falciparum and T. cruzi but also apply to other parasitic protozoa. The described electrophoresis conditions have been developed for the Pulsaphor/Gene Navigator PFGE apparatus (Pharmacia) based on the CHEF (clamped homogeneous electric field) design. Similar results have been obtained with the CHEF-DR system (Bio-Rad). 1.The agarose concentration and type of agarose depends on the DNA size range to be separated. For example, pulsed field separations larger than 2 megabases are improved by using high gel strength agarose (Chromosomal grade agarose, Bio-Rad) allowing preparations of very low percentage agarose gels (0.4-0.6%). 2. For a 15 cm x 15 cm x 0.5 cm 1% agarose gel, dissolve 1.1 g SeaKem GTG agarose in 110 ml 0.5 x TBE buffer by heating in a microwave oven. Make sure that the volume has not changed. Otherwise adjust again to 110 ml. 3. Cool the agarose in a 50 C waterbath and pour into the casting stand. 4. After the gel is solidified, fill the wells with 0.5 x TBE buffer and insert the blocks containing parasite DNA. About 2-5 mm of a block (1-2.5x10^7 parasites) generally contains enough material to visualize the chromosomes of many protozoan parasites (Plasmodium, Leishmania and Trypanosoma) after ethidium bromide staining. However, for best PFGE results it might be necessary to determine for each organism the optimal number of cells /block. 5. Seal the blocks into the well with 1% low melting point agarose. 6. Fill 2.2 l of TBE buffer in the electrophoresis chamber. The buffer should just cover the top of the agarose gel. The temperature of the running buffer (18 C) is generally kept constant during the run. 7. The running conditions which allow the separation of plasmodial and most other protozoan parasite chromosomes in one run (500-4000 kb) are shown in Table. Staining of chromosomes and southern hybridization - 1. After the completion of the run chromosomes can be visualized by staining the agarose gel for 15 min in ethidium bromide (1 mg/ml H2O). The gel is destained by two washes in 0.5xTBE for 1 hr with gentle agitation and photographed (do not forget the ruler) using a shortwave UV-light (254 nm). If chromosomes are intended to be used for subsequent restriction mapping studies longwave UV-light (360 nm) has to be used to avoid nicking of the DNA. 2. For Southern hybridization the DNA is transfered from the agarose gel to a nylon membrane such as Hybond N^+ or BIOTRANS^+. In order to ensure efficient transfer of large DNA fragments the chromosomal DNA has to be nicked. This can be achieved by exposing the stained agarose gel for 5 min on a UV-light table (254 nm). 3. The alkaline transfer procedure using 0.4 N NaOH as transfer buffer works fine for chromosome DNA blots. We routinely set up the capillary transfer (standard molecular biology procedure) for at least 24 hr. Make sure that the weight on top of the absorbent paper stack does not exceed 0.5-1 kg. There is no need to fix DNA after alkali blotting. 4. ^32P-labeld DNA probes using the random hexamer priming method in combination with the hybridization buffer 7% SDS, 0.5 M EDTA pH 8.0, 1% BSA, gives usually a good signal to background ratio with nylon membranes. Single copy genes of P. falciparum are generally detected after 6 hr to overnight exposure. 5. Probes are removed by standard protocols or as recommended by the manufacturer and stripped blots can be stored for prolonged periods at room temperature. Restriction enzyme digestion of embedded chromossomes - Generally, restriction enzymes diffuse into agarose blocks and thus, are suited for chromosome mapping studies. The most useful restriction endonucleases are those with eight base recognition sites which cut only a few times in chromosome sized DNA fragments. However, in genomes which are biased in their AT content, such as P. falciparum, certain enzymes which recognize GC-rich six base pair sequences (SmaI, BglI) can also be used as `rare' cutters in these AT-rich genomes. 1. For restriction enzyme digestion of agarose embedded total parasite DNA, lysis buffer has to be removed by extensive washes in TE buffer (2-3 washes in 20 ml TE). Any remaining proteinase K has to be inactivated by treating the blocks with 1 mM phenylmethylsulphonyl fluoride (PMSF) in TE (10 ml for 20 blocks) for 2 hr at room temperature followed by three washes in 10 ml TE for 30 min each. 2. Individual chromosomes or DNA fragments that have been cut out from a stained agarose gel after a PFGE run must be treated with sec-butanol to extract remaining ethidium bromide prior restriction enzyme digestion. Add 9 vol of sec-butanol saturated in 1M NaCl/TE and 1 vol of the aqueous phase to the gel block (5:1, v/v) and agitate at room temperature for 30 min. Repeat twice the extraction and wash the block five times in 10 ml TE buffer for 30 min each. Store blocks at 4 C. 3. Equilibrate the blocks with 5 vol of restriction buffer containing 100 mg/ml nuclease free BSA for 30-60 min at room temperature. Remove the buffer and add 2 vol of fresh buffer and the restriction endonuclease (approx. 100-200U/ml). Incubate at the recommended temperature for 4 hr or over night. 4. Partial digestion of chromosome DNA is useful for mapping studies. In this case separate digestions of serial dilution of the restriction enzyme (0.01U/ml, 0.1U/ml, 1U/ml, 10U/ml and 100U/ml) can be performed for 2 hr. Reactions will be stopped by adding EDTA to a final concentration of 50mM. 5. Depending on the expected fragment size, restricted DNA will be separated by PFGE according to the run conditions described in Table. Two dimensional PFGE - Most chromosomes of a given protozoan parasite are heterogeneous in size and thus can be separated from each other by PFGE. Two dimensional (2D)-PFGE studies of a karyotype can give useful information concerning genetic markers located on several chromosomes. Multi-gene families are often observed in pathogenic protozoa. A schematic view of a 2D-PFGE is shown in Fig. 2A. An example of a 2D-chromosome blot of P. falciparum is shown in Fig. 2B.
Fig. 2: two dimensional PFGE. A: schematic representation of 2D-PFGE B: Southern-blot of 2D-PFGE analysis of Plasmodium falciparum chromosomes digested with EcoRI. A DNA probe was used that hybridizes to a large gene family dispersed on various chromosomes. The chromosome number is indicated in the top of the gel. 1. Chromosomes are separated in the first dimension and stained with ethidium bromide as described above. 2. The gel is photographed using longwave UV-light (360 nm) and strips of gel (approx. 2 mm) containing all chromosomes are excised with a razor blade. Ethidium bromide is extracted using sec-butanol in a 15 ml Falcon tube. 3. Restriction enzyme digestion is performed as described. 4. The slice of gel containing the digested chromosomes and size markers (lambda ladder or 5 kb ladder) is embedded into a precut slot in the top of a 1.3% agarose gel and sealed with 1% low melting point agarose. 5. Carry out standard PFGE separation using the run conditions described in Table. For Southern hybridization continue as described previously. ACKNOWLEDGEMENTS To Prof. L Pereira da Silva for his support. REFERENCES Copyright 1997 Fundacao Oswaldo Cruz The following images related to this document are available:Photo images[oc97153a.jpg] [oc97153c.jpg] [oc97153b.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}