 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Towards the Physical Map of the Trypanosoma cruzi Nuclear Genome: Construction of YAC and BAC Libraries of the Reference Clone T. cruzi CL-Brener I Ferrari, H Lorenzi, MR Santos*, S Brandariz, JM Requena**, A Schijman, M Vazquez, JF da Silveira*, C Ben-Dov, C Medrano, S Ghio, P Lopez Bergami, I Cano*, B Zingales***, TP Urmenyi****, E Rondinelli****, A Gonzalez*****, A Cortes*****, MC Lopez*****, MC Thomas*****, C Alonso**, JL Ramirez******, MA Chiurrillo******, R Rangel Aldao*******, A Brandao********, W Degrave********, V Perrot, *********, M Saumier*********, A Billaut*********, D Cohen*********, D Le Paslier **********, MJ Levin/^+ Instituto de Investigaciones en Ingenieria Genetica y Biologia Molecular
(INGEBI, CONICET, FCYEN-UBA), Obligado 2490, 1428 Buenos Aires,
Argentina Received 20 August 1997; Accepted 10 September 1997
Strategies to construct the physical map of the Trypanosoma cruzi nuclear genome have to capitalize on three main advantages of the parasite genome, namely (a) its small size, (b) the fact that all chromosomes can be defined, and many of them can be isolated by pulse field gel electrophoresis, and (c) the fact that simple Southern blots of electrophoretic karyotypes can be used to map sequence tagged sites and expressed sequence tags to chromosomal bands. A major drawback to cope with is the complexity of T. cruzi genetics, that hinders the construction of a comprehensive genetic map. As a first step towards physical mapping, we report the construction and partial characterization of a T. cruzi CL-Brener genomic library in yeast artificial chromosomes (YACs) that consists of 2,770 individual YACs with a mean insert size of 365 kb encompassing around 10 genomic equivalents. Two libraries in bacterial artificial chromosomes (BACs) have been constructed, BACI and BACII. Both libraries represent about three genome equivalents. A third BAC library (BAC III) is being constructed. YACs and BACs are invaluable tools for physical mapping. More generally, they have to be considered as a common resource for research in Chagas disease. Key words: Trypanosoma cruzi - genome project - physical map - Yac library - BAC library - sequence tagged sites (STS) - expressed sequence tags (EST) Since the characterization of the first cases of Chagas disease caused by the parasitic protozoan Trypanosoma cruzi (Chagas 1909, Rosenbaum 1964), researchers in Latin America have been attracted to it. During the past 10 years, more than 50 parasite genes have been cloned in our continent, and many immunological features of the disease have been established (Hontebeyrie-Joskowicz Minoprio 1991, Levin 1991, Andrews 1993, Aldao 1993, Mosca Briceno 1993, Gomes 1995, Gonzalez Moro 1995, Pereira-Chioccola et al. 1995, Urbina 1995). With support from national health services and international agencies T. cruzi became a key model organism for laboratories in the region, and was adopted by laboratories in other countries of the continent, and overseas. Therefore, it was inevitable and logical to propose and launch the T. cruzi genome project, as a joint venture between laboratories from Central and South America and laboratories from developed countries. The first step toward the characterization and sequencing of the T. cruzi genome is the construction of its physical map. Strategies to attain this objective have to capitalize on three main advantages of the parasite genome, namely (a) its small size, (b) the fact that all chromosomes can be defined, and many of them can be isolated by pulse field gel electrophoresis (PFGE), and (c) the fact that simple Southern blots of electrophoretic karyotypes can be used to map sequence tagged sites (STS) and expressed sequence tags (EST) to chromosomal bands. A major drawback to cope with is the complexity of T. cruzi genetics, that hinders the construction of a comprehensive genetic map: the parasite reproduces asexually by binary fission, has a clonal population structure, and due to poor chromatin condensation, chromosomes can not be visualized (Solari 1980, Tibayrenc Ayala 1988). An initial experimental strategy for producing a high-resolution physical map of the T. cruzi 87 Mb genome should be based on large genomic fragments cloned in yeast artificial chromosomes (YACs) (Burke et al. 1987, Smith 1994) and bacterial artificial chromosomes (BACs) (Shizuya et al. 1992, Wang 1994), which can be assigned to genomic locations by hibridization with different markers and chromosome specific probes. YACs were chosen because of the large DNA fragments that can be accomodated. The initial reliance on YAC clones was justified because previous works showed that YACs containing T. cruzi genomic fragments were stable and the cloned sequences were colinear with the genomic ones from which they derived (Ajioka Swindle 1993, Don 1995). Moreover, YACs have been successfully used to clone genomic fragments of another protozoan parasite, Plasmodium falciparum. They were employed to disclose the structure of P. falciparum chromosomes (Lanzer 1993, de Bruin 1994). In turn, the P. falciparum genome consortium constructed the YAC library of the project to serve as a common resource in whole genome mapping of this microorganism (The Wellcome Trust Malaria Genome Collaboration 1995). More recently, a cloning system based on BACs has been developed, which appears to offer a reasonable compromise between the technical advantages of cosmids, which harbor insertions of no more than 35 kb, and the larger DNA insert size of YACs. The BAC system is based on the single copy plasmid F factor of Escherichia coli (Shizuya 1992, Wang 1994), that is capable of maintaining foreign DNA fragments of more than 200 kb, with a high degree of structural stability. In fact, the F factor not only codes for genes that are essential to regulate its own replication but also controls its copy number. The pBAC vector incorporates these essential genes as well as a chloramphenicol resistance marker and a multiple-cloning site (Shizuya 1992, Wang 1994). As discussed below, this system has been assigned a primary role in the T. cruzi project. Herein we report the construction and partial characterization of large DNA insert libraries of the T. cruzi nuclear genome in YAC and BAC vectors, which will serve as backbones of the physical map of this complex genome. MATERIALS AND METHODS Trypanosoma cruzi CL-Brener, reference clone of the project - Genomic DNA used to construct the libraries was derived from the reference clone T. cruzi CL-Brener maintained and distributed by Dr B Zingales (USP, Sao Paulo, Brazil; see the corresponding article in this volume). Preparation of T. cruzi CL-Brener genomic DNA - Epimastigotes were grown at the Dept. of Immuno Parasitology of the Pasteur Institut, Paris, France, by Dr Paola Minoprio, and at the Escola Paulista de Medicina, Sao Paulo, Brazil, by Dr J Franco da Silveira. Parasites were harvested, counted and included in an agarose matrix, in order to protect genomic DNA from mechanical rupture (Dausset 1992). Briefly, freshly harvested parasite cells were loaded onto 1% low melting point agarose buffered in PBS (136 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.75 mM KH2PO4) to prepare 100 ul volume plugs containing approximately 20 ug of DNA per plug. Agarose plugs were incubated overnight in NDS (0.5M EDTA, 0.1M NaCl, 1% N-lauroyl- sarcosine pH 8.0) with 1 mg/ml proteinase K (Boehringer Mannheim, Germany) at 50 C and washed several times in 50ml TE (10 mM Tris-HCl pH 7.5, 1 mM EDTA). Construction of YAC libraries Partial digestion of DNA - T. cruzi embedded DNA was partially digested with EcoRI; plugs were preincubated in digestion buffer containing 10 mM MgCl2 and EcoRI was added at a ratio of 5U/ug of DNA and incubated at 37 C for 1hr 30 min. The reaction was stopped by addition of EDTA to a final concentration of 20 mM. DNA size fractionation was followed by analytical PFGE. Preparative PFGE of digested DNA - The partially digested DNA was size-fractionated using a contour clamped homogeneous electric field apparatus (CHEF II, BioRad, California, USA). The gel (0.5x TBE, 0.8% low melting point agarose) was electrophoresed at 140V, for 18 hr at 12 C, with a pulse time of 60 sec. In these conditions, fragments above 400 kb concentrated in the compression zone of the gel. A gel slice containing the compression zone was removed and equilibrated with ligation buffer (see below). Preparation of vector - The vector pYAC4 was linearized by BamHI digestion, and the ends dephosphorylated. After phenol extraction and ethanol precipitation, the pYAC4 cloning site was cleaved with Eco RI. DNA ligations - The agarose slice containing size-selected digested DNA was washed twice in ligation buffer (66 mM Tris-HCl, pH 7.5; 10 mM MgCl2) without ATP (adenosine triphosphate). An 80 fold molar excess of vector was added to the insert DNA. The agarose slice was melted at 68 C for 10 min and loaded in a mold slice apparatus before resolidifying the sample at room temperature. Agarose was then equilibrated in ligase buffer, containing 1 mM ATP and 15U of T4 DNA ligase (Boehringer Mannheim, Germany)/g of gel. Ligation was performed at 4 C for 1 hr and continued afterwards overnight at 14 C. Ligated products were size-fractionated as mentioned above. A slice of agarose containing ligated parasite DNA from the compression zone was removed and equilibrated overnight in agarase buffer before transformation . Transformation of yeast spheroplasts - A colony of yeast strain AB1380 (MATa ade2-1 ura3 can1-100 lys2-1 trp1 his5 psi+) grown onto YPD agar plates (1% yeast extract, 2% bactopeptone, 2% D-glucose, 1.5-2% bactoagar, pH 5.8) was cultured overnight in 200 ml of YPD liquid medium at 30 C to obtain a culture density between 1 to 5 x 10^7 cells/ml. A series of tubes containing 1.5 x 10^9 cells were resuspended in 10 ml SCEM buffer (2M sorbitol, 1M sodium citrate, 50mM EDTA, 14M beta-mercaptoethanol, pH 8.0), prepared and treated with serial dilutions of fresh zymolyase (Seikagaku Corporation, Tokyo, Japan). This treatment was performed at 30 C for 15 min with gentle agitation. Spheroplasts were placed in YPD containing 1M sorbitol. The cells were resuspended in STC buffer (1M sorbitol, 1mM Tris-HCl pH 8.0, 1mM CaCl2). Selected spheroplast preparations revealed an OD600 ratio H2O/sorbitol of 0.5, presenting on microscopic analysis round cells in sorbitol, that were lysed when placed in H2O. Ligated genomic DNA (10 ul per tube) was added to the spheroplast preparation (1.5 x 10^7 cells in 100 ul) for 10 min at room temperature. Then, 1 ml of 20% PEG buffer (PEG 8000, 10 mM Tris-HCl pH 7.5) was added to the mixture for 10 min at room temperature. After centrifugation at 380 g/room temperature, treated spheroplasts were resuspended in 150 ul of SOS buffer (1M sorbitol, 0.25% yeast extract, 0.5% bactopeptone, 20 ug/ml uracil tryptophane, 20 mM CaCl 2, pH 5.8) and incubated at 30 C for 30 min. Thereafter, they were plated on 1.5% top agar containing 1M sorbitol and lacking uracil onto SD Ura^- plates (0.9M sorbitol, 3% D-glucose, 0.67% yeast nitrogen base without amino acids, YNB,1.5-2% bactoagar, pH 5.8 + amino-acids). Plates were incubated at 30 C for at least six days. Red colonies were picked in double selection Ura^- Trp ^- plates. Preparation of DNA for YAC sizing - 100 ul plugs were prepared in low melting point agarose from yeasts grown in overnight cultures at 30 C on YPD. The plugs were incubated for 3 hr with SCEM (1M sorbitol, 50mM EDTA, 0.1M sodium citrate, 7 mM beta-mercaptoethanol) with zymolyase (10 units/plug) at 37 C. Then, they were washed and treated with 1ml PKB (0.1M NaCl, 50mM EDTA pH 8.0, 0.1M Tris-HCl pH 8.0, 1% N-laurylsarcosine with 1mg/ml of proteinase K) at 50 C overnight and finally washed several times with TBE 0.5x to remove proteinase K. Analysis of YAC insert size - YACs were subjected to analytical electrophoresis on 1% low melting point agarose gels in 0.5x TBE by CHEFII migrations with pulse times of 60 to 90 sec., for 18 hr at 12 C, 220v. After migration, the gel was stained with Ethidium Bromide and transferred to a nylon membrane (Hybond-N, Amersham, Buckinghamshire, UK). Membranes were prehybridized and then hybridized at 65 C with ^32P-dCTP labelled T. cruzi total DNA. Preparation of YAC pools - YACs were grown individually in AHC (0.67% YNB, 0.1% casein, 0.002% adenine) for two days at 30 C. Cultures were pooled in plates, arranged in columns and lines, centrifuged at 2000 rpm. Thereafter, they were used to prepare 100 ul plugs with low melting point agarose. Plugs were incubated during 3 hr at 37 C in 10 ml of SCEM/zymolyase (10 units/plug), washed and treated with PKB (0.1 M NaCl, 50 mM EDTA pH 8, 0.1 M Tris-Cl pH 8, 1% N-lauroyl sarcosine with 1 mg/ml proteinase K) at 50 C overnight. The plugs were then washed several times with TBE 0.5% to remove proteinase K. Polymerase chain reaction (PCR) analysis of YAC pools - For PCR assays, plugs containing YAC DNA were treated with agarase (Epicentre Technologies, France); the DNA was diluted in water, boiled for 10 min and finally stored at -70 C. Primers for PCR were designed from DNA sequence data generated in our laboratory: SIRE sense primer 5' GATCGTGGGAGAGCTGGCTA 3', SIRE antisense primer 5' TCCTCCGGGCAGC TGGCCGGATCCTGA 3' (Vazquez 1994); HSP70 -sense primer 5' GGGCACCGGTAAGCGG AACCAG 3' and antisense primer 5' TCCTGA CGAAGACGACGGGTTCG 3' (Levy-Yeyati 1991); gp90 sense primer 5' TATAGGATCCCC TGTGAACACTCGG 3' and antisense primer 5' TTAGTCTAGATT TCCCGTCCTTGAT 3' (Franco 1994); JL7-H49 sense primer 5' GCGGA ATTC ATG GAGCAGGAGCGCAGG 3' and antisense primer 5' GCGGGATCCAACAAA GTGG CTGTCGTC 3' (Levin 1989, Cotrim 1995). PCR reactions were carried out in 50 ul volume reaction mixtures containing 200 uM of each of the deoxyribonucleotide triphosphates (Pharmacia, Uppsala, Sweden), 1x Taq polymerase buffer (Perkin-Elmer, California, USA), 1.25U of Taq polymerase (Perkin Elmer; California, USA) and overlaid with 75 ul of mineral oil (Perkin -Elmer, California, USA). The reaction conditions for each assay were optimized for annealing temperature and for MgCl2 and primer concentrations. An initial denaturation step for 300 sec at 94 C and a final step of 500 sec at 72 C were included, and PCR was performed for 40 cycles. Construction of BAC libraries - Partial digestion of DNA - Genomic T. cruzi DNA embedded in agarose, obtained as described above, was partially digested with Nde II and fractionated by a preparative PFGE (CHEF II). This PFGE was carried out in 1% low melting point at 12 C and 190 v during 18 hr with pulse times of 60 sec. Two libraries were constructed: BAC I, with size selected genomic fragments ranging from 100 to 300 kb and BAC II, with size selected fragments ranging from 200 to 400 kb. Preparation of BAC vectors - The BAC I library was constructed in pBAC108L vector (Shizuya 1992). The BAC II library was constructed in the pBAC derived vector, pBeloBAC11, that allows identification of transformed E. coli cells with blue-white phenotype. Plasmid BAC vectors were linearized with BamHI and dephosphorylated. BAC ligation - 3-5 mm slices of agarose were prepared from size fractionated DNA and equilibrated in TE containing 50 uM NaCl. Thereafter, agarose was melted at 65 C for 15 min, equilibrated at 45 C for 3 min, and incubated 1 hr with agarase (1.5 U/100 ul molten agarose). Finally, the tube was centrifugated to discard undigested agarose and the supernatant kept on ice. For ligation, insert and vector DNAs, in ratios 1:5 and 1:10, were heated briefly and cooled prior to addition of ligase buffer containing 1mM ATP and 1U of T4 DNA ligase in a final volume of 50 ul. Ligation was performed overnight at 16 C. Ligated samples were dialyzed by drop dialysis against TE for 2 hr before electroporation. Preparation of electrocompetent cells - An aliquot from an
overnight culture of E. coli DH10B was incubated in 600 ml of LB at
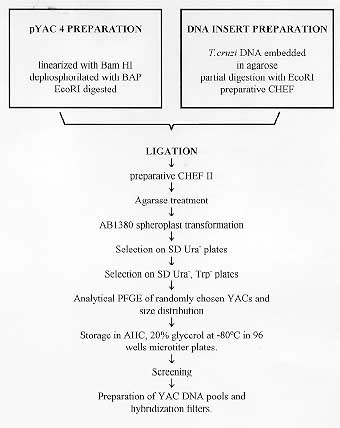
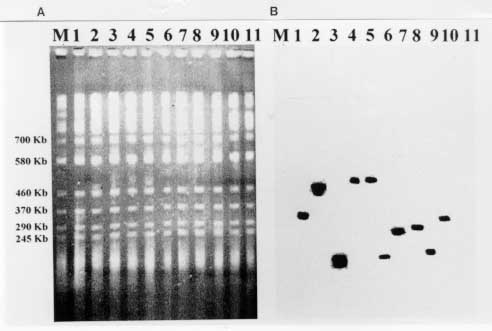
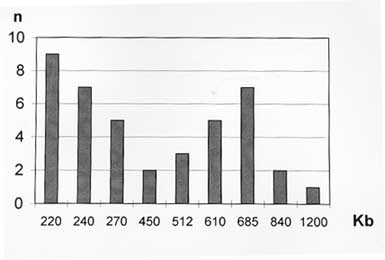
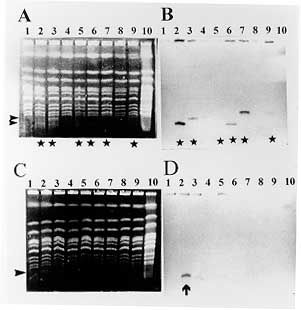
37 C up to an BAC insert sizing - Minipreps were performed by an alkaline lysis method from overnight LB-cloranphenicol cultures (Sambrook 1989). The DNA was digested with 5U of Not I for 4 hr at 37 C. After addition of loading buffer, the samples were electrophoresed in PFGE gels at 6v/cm for 15 hr with pulses of 20/20 sec in a CHEFII apparatus. Storage of libraries - The YAC library was arrayed in 96 wells microplates, growing the cells into YPD and adding 20% glycerol, or in AHC agar microtiter plates, and storage at -70 C. BAC libraries were also arrayed in 96 wells microplates and stored in 20% glycerol after growing them into LB. RESULTS Construction of the YAC library of Trypanosoma cruzi CL-Brener - The basic features of the construction of large insert YAC libraries have already been described (Burke 1987, Dausset 1992). The cloning vector, pYAC4 (Smith 1994), carries sequences necessary for an artificial chromosome: the centromere (CEN4) and an autonomous replicating sequence (ARS). The telomeres were derived from the ciliate eukaryote, Tetrahymena, (Burke 1987). It carries selectable markers on each vector arm (TRP1 and URA3) as well as at the cloning site (SUP4; Burke 1987). In our experiments, the vector was propagated as bacterial plasmid and after modification and ligation to the insert DNA, transformed in yeast host cells as a linear molecule. The host strain AB1380 allowed to take advantage of the selective features of pYAC4 (Burke 1987). Consequently, transformants grew in medium lacking uracil and tryptophan and showed the red phenotype characteristic of the interrupted SUP4tRNA gene (Burke 1987). To apply the YAC cloning technology to the T. cruzi genome, we constructed a YAC library that includes YACs obtained in 4 successful transformations. It is composed of 2,770 clones arrayed in 30 microtiter plates and comprising approximately 10 genome equivalents. To prepare the library it was of outmost importance to obtain intact genomic DNA from epimastigote cells. In a typical experiment, 3 x 10^9 epimastigotes were resuspended in 3 ml of low melting point agarose to prepare aproximately 30 blocks of 100 ul, each containing 20 ug of genomic DNA. Integrity of the embedded genomic DNA was evaluated by PFGE. Indeed, more than 10 chromosomal bands, an intense compression zone and the lack of smears indicated an unaltered molecular karyotype (Cano 1995). Thereafter, conditions for partial EcoRI digestion were tested. The best results were obtained using 5U/ug genomic DNA. To prepare genomic inserts, 100 ug of genomic DNA was partially digested and size fractionated in preparative PFGE (Fig. 1). Fragments above 400 Kb were recovered within the compression zone of the gel and ligated to the pYAC4 vector arms in agarose as described in Material and Methods (Fig. 1). The ligation products were fractionated again by PFGE under similar conditions. After recovery and agarase treatment, size selected ligated DNAs were used to transform yeast spheroplasts. After each transformation, YACs were analyzed for molecular size (Fig. 1, Fig. 2, Fig. 3 ). Fig. 1: YAC cloning procedure. Fig. 2: analysis of sizes of YACs. Eleven randomly selected YAC clones were analyzed by (A) PFGE (conditions of the run are described in Materials and Methods); and (B) Southern blot. The Southern blots were probed with labeled Trypanosoma cruzi total DNA . Fig. 3: size distribution of the Trypasomona cruzi YAC library. Fifty eight randomly chosen YACs were sized by PFGE as described in Materials and Methods; the mean size was calculated to be 365 kb. Library organization and screening- Analysis of 60 randomly selected YACs revealed that the mean insert size was 365 Kb. YACs were organized into an arrayed library in 96 wells microtiter plates. This arrangement offered advantages for screening, including avoidance of loss of slow growing clones and recovery of a maximum number of clones. It enabled also large scale screening using the PCR technique. Two procedures were used to evaluate the representation of different genomic sequences among YACs: one based in the preparation of filters representing 96 wells microplates, the second one based on PCR screening of three dimensional YAC DNA pools (Fig. 4).
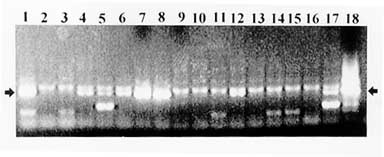
Screening for repetitive sequences - One of the toughest challenges for any cloning system is the stable cloning of repetitive DNA. Interestingly enough, detection of these sequences allows a first evaluation of the representation of the library. Accordingly, we designed a PCR protocol to screen YAC DNA pools for a short interspersed repetitive element of the nuclear genome of T. cruzi, SIRE (Vazquez 1994) distributed in all chromosomes (Levin 1994, Cano 1995). The screening of pools of DNA from rows and columns of a microplate is shown in Fig. 4. In 14 out of 18 pools, SIRE or SIRE-derived amplification products were detected, indicating that for this microplate 55 out of 96 clones carried SIRE or SIRE related sequences. SIRE amplicons were also detected in 7 DNA pools, each one representing 4 microplates that cover the whole library. Screening for gp90 genes - The gp90 gene is present in about 40 copies per haploid genome, and maps to 10 out of 20 chromosomal bands (Cano 1995). PCR 3 dimensional screening of pools covering 200 YACs allowed identification of 12 YACs containing gp90 genes. Confirmation of these results is shown in Fig. 5.
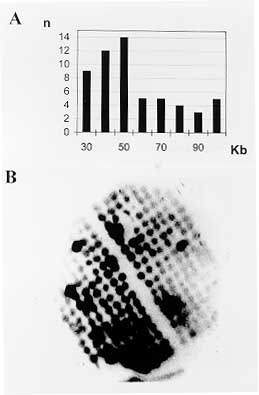
Screening for HSP70 genes - The HSP70 gene locus maps to chromosomal bands IX and X. PCR 3-dimensional screening of 400 YACs identified a YAC containing an HSP70 coding region. Screening for the JL7-H49 genes - Genes coding for antigen JL7-H49 map to 2 homologous chromosomes, XVI and XVII, and define the JL7-H49 locus linked to the JL8 gene (Cano 1995, Cano da Silveira, personal communication). Hybridizations of filters covering 10 microplates permited detection of at least 3 JL7-H49 positive clones. JL7-H49 amplicons of 199 bp were amplified from genomic DNA of the corresponding YACs. These first results indicate that the representation of the library lies within expected ranges, with approximately 60% of clones containing SIRE sequences, about 10% containing gp90 genes, and a low percentage of YACs with JL7-H49 and HSP70 genes. The internal structures of the YAC clones containing gp90, JL7-H49, and the latter HSP70 were examined by comparing the length of restriction fragments from the YAC clones with those of the T. cruzi CL-Brener genomic DNA. In each case, the bands from the YAC clone were of the expected size and matched equivalent bands from the genomic DNA (Lorenzi H, Santos M, da Silverira F, Levin MJ, paper in preparation). In summary, the first experimental tests of the YAC cloning system imply that the studied YACs contain faithful, and genetically stable replicas of the T. cruzi nuclear genome. DNA of YACs do not appear to rearrange, but more detailed tests have to be carried out on a small scale and with a small number of clones in order to exclude the possibility that a certain percentage of YAC clones may be rearranged or unstable (Levin 1994, Brandariz 1995). BAC library construction - Up to date, two types of BAC libraries have been constructed. The initial library of 9,000 clones, BAC I, was made using pBAC108L (Shizuya 1992). To prepare this library, intact genomic DNA was partialy digested with NdeII, size fractionated and ligated to dephosphorilated BamHI digested pBAC (Fig. 6). Ligated pBACs were used to transform competent E. coli cells by electroporation, as described in Materials and Methods. The recombinants were plated, colonies arrayed in 96 wells microplates, and stored for further analysis. A second library consisting of 3,000 clones (BAC II) was constructed using BamHI partially digested genomic DNA. Size selected DNA was recovered from gel using agarase, and ligated. Traditional colony lifts and hybridizations were used for screening and evaluation of insert size (Fig. 6). Representation and stability of BACs from the BAC I library was evaluated. In Fig. 7, the screening of a colony filter with the repetitive sequence E13 is shown. Sizing of the E13 positive BACs revealed a mean BAC insert size of 55kb. However, the mean insert size of 75 randomly selected BACs was not as high, reaching 20kb. Screening of 2,000 BACs with a retroposon probe (Martin 1995) allowed isolation of 32 BACs containing different portions of this element, in agreement with the number of copies of the retroposon in the genome and its chromosomal distribution (Cano 1995). The mean insert size of the BAC II library was slightly higher than that of the first library, 35kb. Several screenings revealed that repetitive elements such as the minisatelite sequence (Gonzalez 1984) were stably inserted. Aiming to increase the insert size of the cloned fragments, a third BAC library (BAC III) is being constructed.
Fig. 7 - A: size distribution of E13 positive BACs from BAC1 library. A total of 59 BACs were analyzed. B: screening of a filter containing 236 BACs with an E13 probe (Requena 1993). DISCUSSION STS are sites in the genome uniquely defined by its DNA sequence. At least three reasons justify their use as primary landmarks in genome mapping (Olson 1989). First, sequences defining STS markers are simple to generate and PCR assays using appropiate primers can be used to identify cloned fragments containing any STS. Second, STS markers can be used to confirm overlaps between clones, since two non chimeric clones containing the same STS must overlap. Third, the definition of STS markers in terms of unique DNA sequences frees the genome map from any particular collection of clones, since the presence of this STS can be assessed in any DNA sample or genomic library. Originally, STS were envisioned as sites chosen at random from anonymous DNA sequences. However, it seemed important to emphasize the use of already cloned sequences as STS markers. An attractive feature of this strategy is that the physical map assembled by means of the STS becomes annotated with biological data, generating valuable knowledge at the earliest phase of construction of the map. Known genes - More than 135 T. cruzi gene sequences have been cloned and sequenced (de Miranda 1995) . Many of them have been mapped to the T. cruzi CL-Brener karyotype obtained by PFGE (Cano 1995, Henrikson 1996) and are actually been used as STS to identify YAC and BAC clones. Furthermore, the characterization of different forms of the same gene allowed to generate additional STS using PCR based single strand conformational analysis (SSCA; Vazquez 1996). SIRE-associated sites (SAS) - An important class of STS markers in T. cruzi consists of sites adjacent to the SIRE. This element is dispersed in the genome and present in approximately 3,000 copies in the CL-Brener genome (Vazquez 1994). The general structure of this sequence is maintained, and can be used to generate STS by inter-SIRE assays, as proposed by Chumakov (1992) and Cohen (1993). More than 20 such SAS are available, and are being mapped to the electrophoretic karyotypes (Lorenzi 1996 ). cDNA sequences or expressed sequence tags - The other important source of STS consists of coding sequences, and are called EST. For our project they are derived mainly from the normalized cDNA library, but other strategies to obtain ESTs are also being used: (a) 5' ESTs are being generated by PCR using different 3' primers and a common sense mini-exon primer (Gonzalez personal communication) and (b) antigenic ESTs are being obtained by massive immunological screening of different lambda expression libraries (Fig. 8; Ghio Levin, unpublished results).
Standardized electroforetic karyotypes: an additional support for physical mapping - To establish the arrangements of all cloned fragments in a continuous stretch of DNA, it is of paramount importance to determine the chromosomal distribution of all markers generated within the framework of the T. cruzi genome project. This type of mapping has proved to be useful in defining linkage groups for the CL-Brener genome (Cano 1995, Henrikson 1996). Comparison of this STS distribution with the organization of the same markers in other strains is being used to determine which markers and/or cloned fragments consistently co-segregate on the same band, regardless of the strain. It is logical that, the more frequently markers co-segregate, the closer they must be. This particular type of genetic analysis is allowing the definition of linkage groups of markers and/or cloned genomic fragments, a basic complement and necessary support for construction of the physical map. Chromosome specific contig assembly, or how to build the map - The first step in building the map of the T. cruzi genome is the use of STS markers as probes to isolate YACs and BACs. Each set of clones identified by a chromosome specific STS forms a contig, since they all contain the same chromosome specific sequence. In our case, contig assembly has begun around several well characterized loci, such as JL7-H49-JL8, and B11-B12-B13. Simultaneously, a modified version of the genome sampling strategy (GSS) (Smith 1994) has been developed. Isolated chromosomes XIX and XX. (Cano 1995) are being used as probes to hybridize high density filter membranes to organize YACs and BACs in chromosome specific subsets. Chromosome specific markers, such as those obtained by inter-SIRE PCR, SAS, are being used to confirm the chromosome specificity of the cloned genomic fragments (Lorenzi 1996). Since BACs are easy to manipulate and cloned DNA is easy to prepare, the first effort concerning the assembly of cloned fragments to establish contigs is based on BACs. Each group of chromosome specific BACs, identified by a particular STS, forms a contig, since they all contain the same STS. End specific probes will be used to extend contig assembly. Ideally YACs will be used to connect established BAC contigs to yield a continuous physical map of each chromosome. We conclude that large insert libraries in YAC and BAC vectors are now available. They are invaluable tools for the construction of the physical map of this complex genome. More generally, they have to be considered as a common resource for research in Chagas disease. This work was supported by: Project Genome Trypanosoma cruzi, INGEBI-CEPH, Ministere d'Affaires Etrangeres, France - Ambassade de France en Argentine; Trypanosoma cruzi Genome Project-Subprograma III-Biotecnologia - CYTED Programa Iberoamericano de Ciencia y Tecnologia para el Desarrollo; the United Nations Development Program for Research and Training in Tropical Diseases (WHO/TDR); INSERM-CONICET cooperation program; Trypanosoma cruzi Genome Project associated to the Human Genome Project of the University of Buenos Aires (Ex203-Ex283SECyT-UBA); Fundacion Antorchas, and European Commission Contract No. 936018 AR.; CNPq/PADCT, Brazil; FAPESP, Brazil. REFERENCES Copyright 1997 Fundacao Oswaldo Cruz The following images related to this document are available:Photo images[oc97157b.jpg] [oc97157a.jpg] [oc97157e.jpg] [oc97157h.jpg] [oc97157c.jpg] [oc97157f.jpg] [oc97157d.jpg] [oc97157g.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}