 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
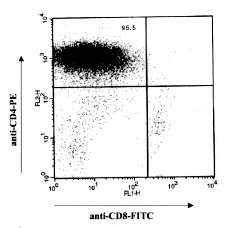
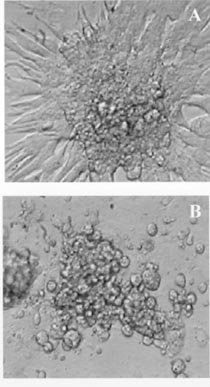
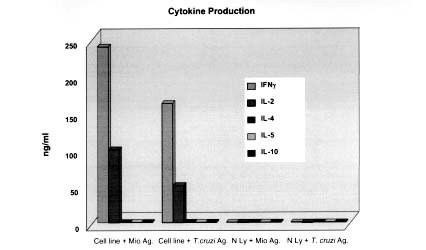
Mem Inst Oswaldo Cruz, Rio de Janeiro, Vol. 94, Suppl. I: pp. 257-262, 1999 Immunopathology of Cardiomyopathy in the Experimental Chagas Disease Milena BP Soares, Ricardo RibeirodosSantos+Laboratório de Imunofarmacologia, Centro de Pesquisas Gonçalo Moniz-Fiocruz, Rua Valdemar Falcão 121, 40295-001 Salvador, BA, Brasil +Corresponding author. Received 9 June 1999 Code Number:OC99174 The mechanisms by which Trypanosoma cruzi causes cardiomyopathy and induces neuronal destruction are discussed in this paper. The results suggest that autoimmunity in the chronic phase is the main cause of the progressive cardiac destruction, and that autoreactivity is restricted to the CD4+ T cell compartment. During the acute phase, the neuronal and cardiac fiber destruction occurs when ruptured parasite nests release T. cruzi antigens that bind to the cell surface in the vicinity which become targets for the cellular and humoral immune response against T. cruzi. The various factors involved in the genesis of autoimmunity in chronic T.cruzi infection include molecular mimicry, presentation of self-antigens and imbalance of immune regulation. Key words: cardiomyopathy - immunopathology - Chagas disease - Trypanosoma cruzi Chagas disease is caused by a protozoan parasite, Trypanosoma cruzi, which is transmitted by reduviid bugs. Approximately 35 million people are exposed to the risk of infection (Puffer & Griffith 1967) and the disease is one of the leading causes of death in many countries of Latin America. The disease is characterized by two major clinical forms: acute and chronic (Ribeiro dos Santos et al. 1981). The acute phase progresses with parasitemia, and the parasites spread widely in the host organism, replicating inside macrophages and in a variety of other host cells with a preference for heart muscle cells. It is characterized histopathologically by foci of myocytolytic necrosis and an impressive mononuclear inflammatory infiltrate directly related to tissue parasitism. The resulting inflammatory destruction is usually of only transitory clinical relevance. Progression from the acute to chronic form of Chagas disease coincides with clearance of parasites from bloodstream and tissues. This period can last up to 20 years, and is called indeterminate phase when no clinical symptoms or significative histopathological lesions are found. The typical chronic form affects mainly the heart and occurs in approximately 25% of the cases. Variable degrees of cardiac hypertrophy and dilatation are found with or without thinning of the apical region (apical aneurysm). Foci of myocardial necrosis and degeneration are present with an inflammatory infiltrate predominantly composed of mononuclear cells and interstitial fibrosis. Importantly, myofibers containing parasites are rarely observed (Köberle 1968). The other clinical forms consists of the megamorphic dilatations of the oesophagus and colon, which occur in less than 10% of the infected individuals, and are mainly the result of neuronal destruction of the gastrointestinal tract (Köberle et al. 1983). The pathogenesis of Chagas heart disease is still not well understood. The host immune response to T. cruzi has predominantly been studied in experimental infections of mice. In the acute phase, the neuronal and myocardial lesions are related to the tissue parasitism. The rupture of the parasite nests results in the release of T. cruzi antigens that sensitize and transform neurons and muscle fibers (Ribeiro dos Santos & Hudson 1980a, Araujo 1985), leading to the destruction of these cells by the immune response against the parasite (Kuhn & Mumane 1977, Ribeiro dos Santos & Hudson 1980b). Recently, in immunofluorescence studies, we have shown binding of monoclonal antibodies against T. cruzi in neuronal cell line (Neuro 2A) incubated with T. cruzi extract, (Fig. 1) indicating that different parasite antigens interact with host cells (manuscript in preparation). Concerning the chronic phase, different mechanisms have been proposed. It has been suggested that the lesions result from a tenacious parasitic invasion. However, the low levels of parasitemia and the very rare finding of tissue parasites strongly argue against this hypothesis (Köberle 1968). Second, microvascular abnormalities have been postulated to play a significant role in the pathogenesis of chronic Chagas heart disease (Rossi et al. 1984, Rossi 1990). Third, the scarcity of parasites, the polyclonal activation in the acute phase and the presence of autoreactive antibodies and T cells directed to target organs are evidence that sustain an autoimmune mechanism in the genesis of chronic disease (Ribeiro dos Santos et al. 1979, Snary et al. 1983, Kierszenbaum 1985, Minoprio et al. 1986, Minoprio et al. 1988, 1989, McCormick & Rowland 1989, Bonfá et al. 1993). We have demonstrated that mice chronically infected with T. cruzi reject syngeneic heart grafts (Ribeiro dos Santos et al. 1992). This was in striking contrast to hearts grafted into normal or T.cruzi-immunized syngeneic recipients, which are not rejected and can persist for more than six months. The study of heart tissue grafted into chagasic mice revealed a persistent and intense mononuclear inflammatory infiltrate, quite similar to the pattern obtained in the allogenic conditions, suggesting that cellular mechanisms are implicated in the rejection. These studies have also shown that only splenic CD4+ from chronically infected mice are able to mediate syngeneic heart graft destruction when injected insitu, whereas CD8+ or non-T cells were not effective. Kinetic studies transferring CD4+ cells show that the autoreactivity starts 15 days after infection and is fully established at the 30th day (Ribeiro dos Santos et al. 1991). To further investigate the phenotype of the cells involved in the phenomenon of syngeneic heart rejection, mice chronically infected with the Colombian strain of T.cruzi were treated with anti-CD4 or anti-CD8 monoclonal antibodies (mAb) before the transplantation. The in vivo depletion of CD4+ cells, but not CD8+ cells, abrogates rejection Interestingly, when implanted two months after the end of the treatment with anti-CD4 antibody, the grafts were not rejected despite the fact that CD4+ population was already restored to normal levels. Instead, these grafts persisted for more than six months, suggesting that the recipients became tolerant, and that this was not a graft adaptation phenomenon. In contrast, allogeneic grafts were not accepted and were rejected in 10 to 22 days post-transplantation. The self-reactivity of CD4+ T cells from chronically infected mice could also be shown invitro since lymphocytes from chronic infected mice proliferate in the presence of heart antigens plus irradiated feeder cells. Non-T cell populations or CD8+ T Iymphocytes failed to show any activity in this assay. Tolerance after anti-CD4 treatment also correlated with the disappearance of myocardial reactivities invitro (Ribeiro dos Santos et al. 1992). In order to investigate the state of tolerance induced by anti-CD4 treatment, we established a model of chronic carditis by infecting mice with 102 Colombian strain trypomastigotes. After 30 days of infection, 60 to 70% of the animals survive to the acute phase of the infection, and enter the chronic phase with low or no parasitemia. Hearts from these animals show multifocal carditis composed of mononuclear cells and the occasional finding of parasite nests in myocardial cells. Approximately 40% of the infiltrating cells are T lymphocytes, with a predominance of the CD4+ phenotype. CD4+ cells were usually spread throughout the inflammatory foci while CD8+ cells have the tendency to be in close contact with parasitized myocardial fibers. Chronic chagasic animals were then submitted to different schedules of monoclonal antibody treatment: (1)anti-CD4; (2)anti-CD8; (3)anti-Thy1. After five months of follow-up, the animals were sacrificed. The histopathological and immunocytochemical analysis of the hearts showed that the chronic carditis was abolished in the animals treated with anti-CD4. The cumulative mortality is very low in these animals, and parasitemia remains negative during the whole period of observation. This is in striking contrast with the mortality rates found with the opposite treatment (anti-CD8 or anti-Thy1), where 90% of the animals die. The few remaining animals showed an intense carditis, two thirds of the inflammatory T cells having a CD4+ phenotype with the majority in close contact with degenerated myocytes (Pirmez & Ribeiro dos Santos 1994). These results establish that autoreactivity is restricted to the CD4+ T cell compartment, clearly different from allogeneic skin graft rejection, which has been attributed to both subsets of T Iymphocytes (Cobbold et al. 1984, Kitagawa et al. 1990). Altogether, these results are consistent with the heart graft model and other experimental models of organ-specific autoimmune diseases in which CD4+ T cells play a major role in the induction of tissue lesions (Sakaguchi & Sagakuchi 1988, 1989, 1990). This is also in keeping with data showing that T cells mediating cellular immunity in experimental T.cruzi infection correspond to CD4+ T cells (Hontebeyrie-Joskowicz et al. 1987, Ben Younes-Chennoufi et al. 1988). Although some experimental models (Tarleton 1991) as well as human studies (Molina & Kierszenbaum 1987, Reis et al. 1993, Cunha-Neto 1994) have shown a predominance of CD8+ cells in heart lesions, it is noteworthy that most of the T-cell lines or clones reacting against T.cruzi antigens or self-components such as myosin obtained so far are CD4+ (Britten & Hudson 1985, Dutra et al. 1992,Nickell et al. 1987) and only occasionally CD8+ (Nickell et al. 1993). To further study the role of autoreactivity in the pathogenesis of the myocardium lesions in Chagas' disease, we have generated a CD4+ T cell line by repeated in vitro antigenic stimulation of purified splenic CD4+ T lymphocytes from chronically T. cruzi-infected DBA mice (Colombian strain) (Fig. 2). These T cells proliferate in the presence of soluble heart antigens and syngeneic feeder cells or in co-cultures with irradiated splenic syngeneic feeder cells and fetal heart cells. The lymphocytes originating the cell line appear to have resulted from the in vivo expansion of T. cruzi-reactive lymphocytes, since the line was activated in vitro by T. cruzi lysates, in addition to heart antigens. The cell line could also decrease the number of beating fetal heart cell-clusters in vitro when co-cultured with irradiated splenic syngeneic feeder cells and fetal heart cells (Fig. 3). In vitro antigen stimulation of the cell line showed a Th1 cytokine profile, with production of high levels of IFN-g and IL-2 and absence IL-4, IL-5 or IL-10 (Fig. 4). In addition, in situ injection of these cells into well established heart transplants induce the cessation of heart beating. Adoptive transfer of the cells to BALB/c nude mice caused 100% mortality of recipients after 1-2 months, compared to controls which received normal CD4+ T cells. Histological studies revealed the presence of multifocal mononuclear infiltrates in their hearts similar to those observed during the chronic phase of T. cruzi infection. No significant alterations were observed in the hearts of BALB/c nude mice transferred with normal splenic CD4+ T cells. This T cell line also induces destruction of neuronal cells (in the presence of feeder cells), but only if the target is previously incubated with parasite antigens (manuscript in preparation). Recently, we have also obtained two T cell lines from BALB/c mice infected for one year with the Colombian strain of T. cruzi. We are currently performing the characterization of the different T cell clones present in these cell lines and of the antigens recognized by them. This should allow us to show the existence of cross-reactive T cells against T. cruzi and heart antigens. Four T cell hybridomas were already obtained by fusion of the first cell line with BW (thymoma) cell line. From these, two showed reactivity against both T. cruzi and heart antigens, one reacted only against parasite antigens, and the last showed reactivity only to heart antigen. The autoreactivity could be theoretically elicited by altered self-antigens, formed during heart inflammation, and/or by T.cruzi antigens cross-reacting with self. In order to identify cross-reactive antigens, we have generated monoclonal antibodies against T. cruzi. Four of them (TC1, TC2, TC8, and TC10) are cross-reactive with myocardium, and each recognises a specific heart and T. cruzi antigen, as shown by western blotting and by immunofluorescence techniques. In conclusion, studies in the murine model of infection with Colombian strain of T. cruzi suggest that the heart lesions may be caused by two different mechanisms. In the acute phase, parasite antigens adsorbed or presented in host cell surface are targets for the anti-T. cruzi immune response, which will lead to destruction of cardiac fibbers and neurones. Lesions are therefore strongly associated with tissue parasitism. Both CD4+ and CD8+ T cells may play a role in the pathogenic response. Release of self-antigens from damaged cells may also contribute to the pathology. Second, an autoreactive response against heart antigens mediated by CD4+ T cells induces injury in the heart. Damage is dependent on accessory cells such as macrophages, possibly activated by IFN-g and other cytokines. The heart-specific autoreactive response present during the chronic phase of the T.cruzi infection may be intensified or even triggered by multiple parasite-host cell cross-reactivities. As the disease progresses driven by an autoimmune response, little association between damage and presence of parasites is observed. In both mechanisms, it is clear that multiple antigens from the parasite as well as self-antigens are targets for the disease promoting cells. ACKNOWLEDGMENTS To Drs José Orivaldo Mengel and Edilberto Postol for the collaboration in Tcell and monoclonal antibodies techniques. REFERENCES Araujo FG 1985. Trypanosoma cruzi: Expression of antigens on the membrane surface of parasitized cells. J Immunol 135: 4149-4154. Ben Younes-Chennoufi A, Said G, Eisen H, Durand A, Hontebeyrie-Joskowicz M 1988. Cellular immunity to Trypanosoma cruzi is mediated by helper T cells CD4+. Trans R Soc Trop Med Hyg 82: 84-89. Bonfá E, Viana W, Barreto ACP, Yoshinari N, Cossermelli W 1993. Autoantibodies in Chagas disease: an antibody cross-reactive with human and Trypanosoma cruzi ribosomal proteins. J Immunol 150: 3817-3923. Britten V, Hudson L 1985. Isolation of human T-cell lines from a patient with Chagas disease. Lancet 21: 637-639. Cobbold DP, Jayasuriya A, Nash A, Prospero TD, Waldmann H 1984. Therapy with monoclonal antibodies by elimination of T-cell subsets in vivo. Nature 312: 548-551. Cunha-Neto E, Durant M, Gruber A, Zingales B, Messias I, Stolf N, Bellotti G, Pillegi F, Kalil J 1995. Autoimmunity in Chagas cardiopathy: biological relevance of a cardiac myosin specific epitope cross-reactive to immunodominant T. cruzi antigens Proc Natl Acad Sci USA 92: 3541-5. Dutra WO, Colley DG, Cançado JR, Brener Z, Martins-Filho OA, Gazzinelli G, Parra JC 1992. T-cell activation and regulation in human Chagas disease: a phenotypic analysis. Mem Inst Oswaldo Cruz 87: 46-47. Hontebeyrie-Joskowicz M, Said G, Millon G, Marchal G, Eisen H 1987. L3T4+ cells able to mediate parasite-specific delayed-type hypersensitivity play a role in the pathology of experimental Chagas disease. Eur J Immunol 17: 1027-1033. Köberle F 1968. Chagas disease and Chagas syndromes: the pathology of American trypanosomiasis. Adv Parasitol 6: 63-116. Köberle F, Alcântara FG, Ribeiro dos Santos R 1983. Patogenia da forma digestiva p. 25-34. In A Raia, Manifestações Digestivas da Moléstia de Chagas, Sarvier. Kierszenbaum F 1985. Is there autoimmunity in Chagas disease? Parasitol Today 1: 4-6. Kitagawa S, Sato S, Hori S, Hamaoka T, Fujiwara H 1990. Induction of anti-allo-class I H-2 tolerance by inactivation of CD8+ helper T cells, and reversal of tolerance through introduction of third-party helper T cells. J Exp Med 172: 105-113. Kuhn RE, Mumane JE 1977. Trypanosoma cruzi: immune destruction of parasitized mouse fibroblasts in vitro. Exp Parasitol 41: 66-73. McCormick TS, Rowland, EC 1989. Trypanosomacruzi: cross-reactive anti-heart autoantibodies produced during infection in mice. Exp Parasitol 69: 393-401. Minoprio P, Eisen H, Forni L, D'Imperio Lima MR, Joskowicz M, Coutinho A 1986. Polyclonal lymphocyte responses to murine Trypanosomacruzi infection. I. Quantitation of both T and B responses. Scand J Immunol 24: 661-668. Minoprio P, Burle O, Pereira P, Guilbert B, Hontebeyrie-Joskowicz M, Coutinho A 1988. Most B cells in acute Trypanosomacruzi infection lack parasite specificity. Scand J Immunol 28: 553-559. Minoprio P, Andrade L, Lembezat MP, Ozaki LS, Coutinho A 1989. Indiscriminate representation of VH-gene families in the murine B lymphocyte responses to Trypanosomacruzi. J Immunol 142: 4017-4021. Molina HÁ, Kierszenbaum F 1987. A study of human myocardial tissue in Chagas disease: distribution and frequency of inflammatory cell types. Int J Parasitol 17: 1297-1305. Nickell SP, Gebremichael A, Hoff R, Boyer MH 1987. Isolation and functional characterization of murine T-cell lines and clones specific for the protozoan parasite Trypanosomacruzi. J Immunol 138: 914-921. Pirmez C, Ribeiro dos Santos R 1994. Autoreactivity in chronic experimental Trypanosoma cruzi infection. Cienc Cult 46: 418-423. Puffer RR, Griffth GW 1967. Chagas disease, p. 48-57. In Patterns of Urban Mortality, PAHO, Washington. Reis DD, Jones EM, Tostes SJ, Lopes ER, Gazzinelli G, Colley DG, McCurley TL 1993. Characterization of inflammatory infiltrates in chronic chagasic myocardial lesions: presence of tumor necrosis factor-a+ cells and dominance of granzyme A+, CD8+ lymphocytes. Am J Trop Med Hyg 48: 637-644. Ribeiro dos Santos R, Marquez JO, Furtado CCG, Oliveira JCR, Martins AR, Köberle F 1979. Antibodies against neurons in chronic Chagas disease. Tropenmed Parasit 30: 19-23. Ribeiro dos Santos R, Hudson L 1980a. Trypanosoma cruzi: binding of parasite antigens to mammalian cell membranes. Parasite Immunol 2: 1-10. Ribeiro dos Santos R, Hudson L 1980b. Trypanosoma cruzi: immunological consequences of parasite modification of host cells. Clin Exp Immunol 40: 36-41. Ribeiro dos Santos R, A Rassi, Köberle F 1981. Chagas disease, p.115. In H Schonfeld & S Karger (eds), Antiparasitic Chemotherapy, Cyba Found. Symp., Elsevier-Experta Medica. Ribeiro dos Santos R, Pirmez C, Savino W 1991. Role of autoreactive immunological mechanisms in chagasic carditis. Res Immunol 142: 134-137. Ribeiro dos Santos R, Rossi MA, Laus JL, Santana-Silva J, Savino W, Mengel J 1992. Anti-CD4 abrogates rejection and reestablishes long-term tolerance to syngeneic newborn hearts grafted in mice chronically infected with Trypanosoma cruzi. J Exp Med 175: 29-39. Rossi MA, Gonçalves S, Ribeiro dos Santos R 1984. Experimental Trypanosoma cruzi cardiomyopathy in Balb/c mice: the potential role of intravascular platelet aggregation in its genesis. Am J Pathol 114: 209-216. Rossi MA 1990. Microvascular changes as a cause of chronic cardiomyopathy in Chagas disease. Am Heart J 120: 233-236. Sakaguchi S, Sagakuchi N 1988. Thymus and autoimmunity: transplantation of the thymus from cyclosporin A-treated mice causes organ-specific autoimmune disease in athymic nude mice. J Exp Med 167: 1497-1485. Sakaguchi S, Sagakuchi N 1989. Organ-specific autoimmune disease induced in mice by elimination of T cell subsets.V. Neonatal administration of cyclosporin A causes autoimmune disease. J Immunol 142: 471-480. Sakaguchi S, Sagakuchi N 1990. Thymus and autoimmunity: capacity of the normal thymus to produce pathogenic self-reactive T cells and conditions required for their induction of autoimmune disease. J Exp Med 172: 537-545. Snary D, Flint JE, Wood JN, Scott MT, Chapman MD, Dodd J, Jessel TM, Miles MA 1983. A monoclonal antibody with specificity for Trypanosoma cruzi, central and peripheral neurons and glia. Clin Exp Immunol 54: 617-624. Tarleton RL 1991. The role of T cell subpopulations in experimental Chagas disease. Res Immunol 142: 130-133. Copyright 1999 Fundacao Oswaldo Cruz - Fiocruz The following images related to this document are available:Photo images[oc99174d.jpg] [oc99174b.jpg] [oc99174c.jpg] [oc99174a.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}