 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Brazilian Journal of Oral Sciences, Vol. 4, No. 13, April./June. 2005, pp. 695-706 Hypodontia: genetics and future perspectives Trevor J Pemberton1 Parimal Das3 Pragna I Patel1,2 1Institute for Genetic Medicine and the 2Center for Craniofacial Molecular Biology, Keck School of Medicine, University of Southern California, CA-USA and 3Department of Surgical Oncology, M.D. Anderson Cancer Center, Houston, TX-USA Correspondence to:Pragna I Patel Institute for Genetic Medicine, Keck School of Medicine, University Of Southern California, 2250 Alcazar Street, CSC-240, Los Angeles, CA 90033, USA. Tel: +1 (323) 442 2751 Fax: +1 (323) 442 2764 E-mail: pragna@usc.edu Received for publication: February 14, 2005 Code Number: os05011 Abstract Tooth development is a complex process of reciprocal interactions that we have only recently begun to understand. With the large number of genes involved in the odontogenic process, the opportunity for mutations to disrupt this process is high. Tooth agenesis (hypodontia) is the most common craniofacial malformation with patients missing anywhere from one tooth to their entire dentition. Hypodontia can occur in association with other developmental anomalies (syndromic) or as an isolated condition (non-syndromic). Recent advances in genetic techniques have allowed us to begin understanding the genetic processes that underlie the odontogenic process and to identify the mechanisms responsible for tooth agenesis. Thus far two genes have been identified by mutational analysis as the major causes of non-syndromic hypodontia; PAX9 and MSX1. Haploinsufficiency of either has been observed to cause the more severe forms of hypodontia whilst point mutations cause hypodontia to varying degrees of severity. With the prevalence of hypodontia having been observed to have increased during the 20th century, the future identification and analysis of its genetic basis is essential to allow us to better treat the condition. The clinician can facilitate this process by collaborating with the human geneticist and referring patients/families with familial hypodontia for investigative research. Key Words: hypodontia, tooth agenesis, PAX9, MSX1, prevalence, mutations The smile is a unique facial expression distinct to primates. The main physical component of the smile is a complete dentition set comprising four different types of teeth. Vertebrate comparative histology has indicated that the continued evolution of teeth throughout the emergence of modern man is due to the increased fitness they have offered us. Our modern lifestyle also has a special attachment to a complete set of dentition aside from their use as merely mastigatory appendages for food. For this reason, naturalists, biologists and dentists have for a long time been trying to unravel the cause for the congenital loss of teeth leading to several clinical phenotypes such as hypodontia, oligodontia and anodontia. While a number of clinical studies have been carried out on disorders that involve the congenital lack of teeth1-11, until recently very little effort has been made to understand the genetic component responsible for mammalian tooth development. Advancements in molecular biology approaches coupled with the now complete human genome sequence12 has allowed a number of putative disease genes/ loci associated with the hypodontia/oligodontia phenotypes to be identified6,13-16. Functional studies of these disease genes have started to reveal their precise role in tooth development allowing us to better understand their role in disease pathology and the molecular morphogenetic fields within which they function17. The congenital lack of one or more permanent teeth is a common anomaly in man. By definition, congenitally missing teeth are those that fail to erupt in the oral cavity and remain invisible in a radiograph, which implies that this is caused by disturbances during the early stages of tooth development. These phenotypes most frequently involve the second premolars and upper lateral incisors and commonly lead to mild phenotypes that have no associated systemic disorder. The term hypodontia has most frequently been used for describing the phenomenon of congenitally missing teeth, although a large number of missing teeth is defined as oligodontia and the complete absence of teeth is defined as anodontia. Hypodontia and oligodontia are also classified as either nonsyndromic (isolated) or syndromic (associated with their syndromes). A literature survey shows various other terminologies describing a reduction in teeth number; teeth aplasia, congenitally missing teeth, absence of teeth, agenesis of teeth, and lack of teeth. Although no distinct definition and classification exists in literature, the following definitions have been widely used in scientific literature:
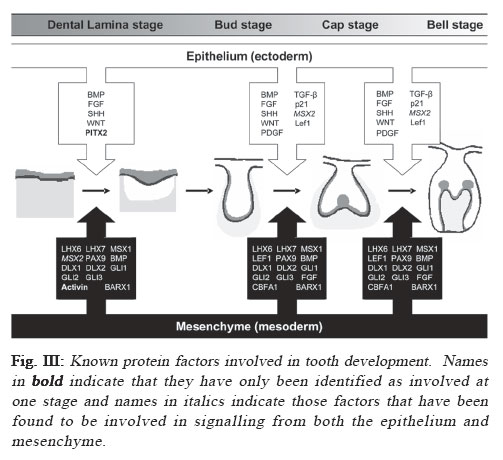
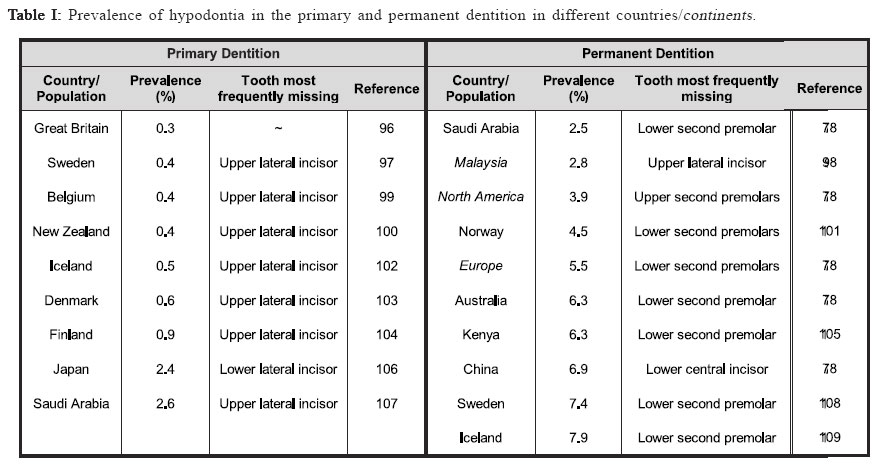
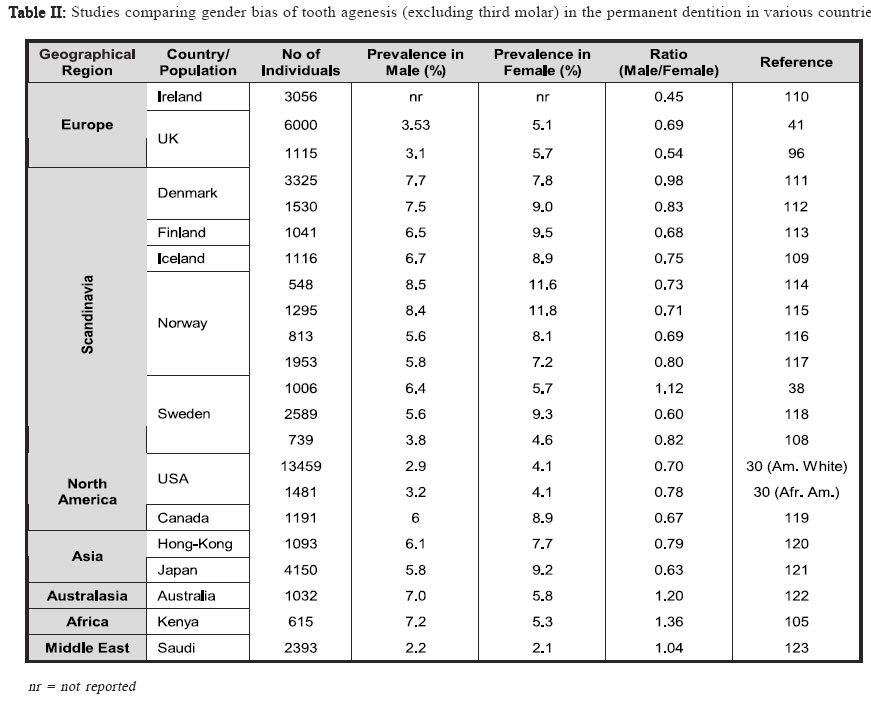
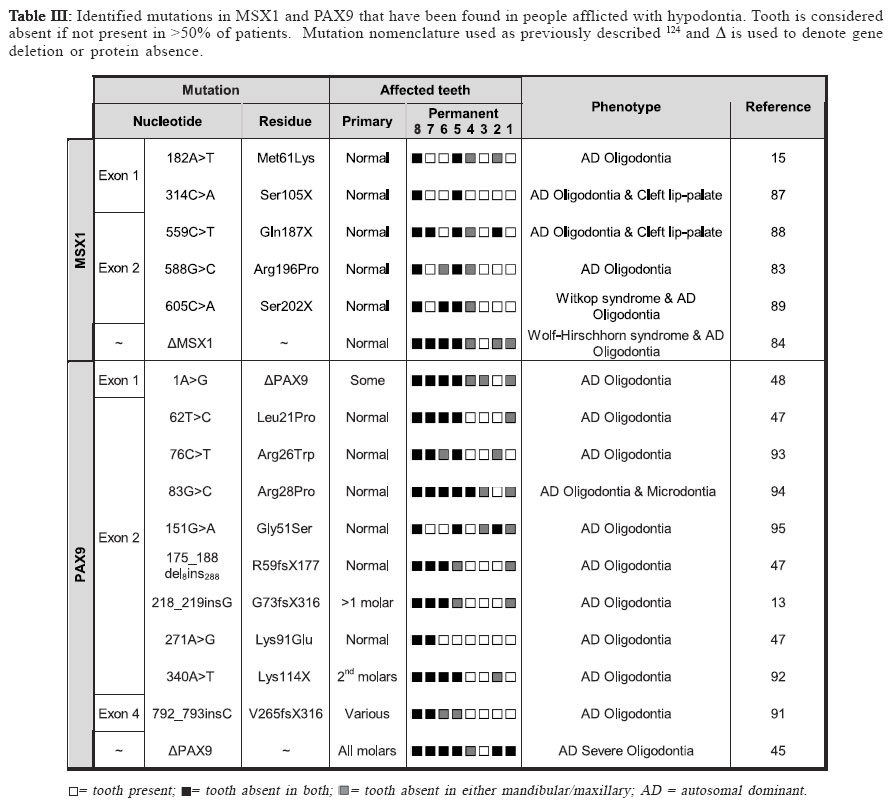
Identifying disease genes The online catalog of inherited human diseases (Online Mendelian Inheritance in Man; OMIM18) currently lists almost 16000 inherited human disease genes. However, the molecular etiology for only about 2000 of these has been determined successfully. The cloning of disease genes is the first step in understanding the detailed molecular basis of the disease that ultimately facilitates the development of suitable diagnostic and therapeutic agents. There are mainly two different strategies that have been employed for identifying human disease genes: functional cloning and positional cloning (Figure I). As the name implies, functional cloning identifies genes based on the known biochemical function of their encoded proteins. For example, the identification of the gene responsible for phenylketonuria was achieved by purifying the mRNA for phenylalanine hydroxylase, the enzyme normally responsible for the oxidation of phenylalanine to tyrosine but absent in sufferers, and using this to screen a cDNA library for the corresponding DNA sequence19 which facilitated the identification of its chromosomal location20-21. However, in reality for the vast majority of inherited disorders, knowledge about their basic biochemical defect is unknown making the use of functional cloning impossible which has led to a second strategy, positional cloning. The positional cloning approach employs one or both of the following strategies; (a) analysis of genetic linkage in families with a disease and/or (b) identification of a specific chromosomal aberration(s) in the diseased individual. Successful genetic analysis depends on the following requirements; (a) identification of large families segregating the disease phenotype, (b) a distinctive diagnostic criterion to distinguish the affected individuals, (c) accurate assessment of the individuals of the family for a known Mendelian or complex pattern of segregation and (d) highly polymorphic DNA markers. In principle, the basis of linkage analysis is to observe the closeness of two alleles for two different genes on a chromosome. The closer the two alleles are to each other, the more likely they are to segregate together during meiosis due to the reduced chance of a recombinatorial event occurring between them. Logically, this principle is applied to determine the genetic distance of known genetic markers, or “site locators”, from a disease locus by tracing its segregation pattern in affected families. In figure II, the disease locus D is closer to the marker locus represented by allele X/x but distant from the locus represented by Y/y. Due to this, the disease locus co-segregates with the X locus whereas it shows random segregation pattern with respect to the Y locus. Essentially, good genetic markers are those which are polymorphic and can be easily assayed in genomic DNA isolated either from lymphocytes or buccal cells. The traditional definition of polymorphism is a natural variation in nucleotide sequence which is observed in at least 1% of a population and is capable of distinguishing the parental chromosomes which otherwise seem virtually identical in every respect. The degree of polymorphism of the genetic marker has direct bearings in scoring the distinction. The information on segregation of polymorphic markers with respect to the disease phenotype is then analyzed using statistical programs in order to derive a LOD score. LOD score, or the logarithm to the base 10 of the odds, is defined as the relative probability of the observed data being the result of true linkage versus the probability due to chance. Conventionally, linkage is considered established if the LOD score at any given recombination fraction (θ) is equal to or greater than 3, while linkage is excluded if the LOD score is equal to or less than –2. Statistically, a LOD score of 3 means that it is 103, or 1000 times more likely that the observed pattern of segregation of one marker with respect to another marker or disease locus occurred because of linkage rather than chance. Multipoint linkage analysis for determining the disease gene position with respect to a number of genetic markers in the candidate region is then used to localize the position of the disease gene between two flanking genetic markers. The success of positional cloning efforts requires very tightly linked markers. An alternative approach to associate gene(s) with a genetic disorder is to analyze mutations in candidate genes, those that have either a proven or speculated association either directly or indirectly leading to the disease state. With the advancement of our present day knowledge about disease pathology and its consequences at both the biochemical and physiological levels, mapping human disease genes by the candidate gene approach plays an important role especially in excluding the involvement of a gene for a given phenotype. Using this approach, the association of multiple growth factor and growth factor receptor gene(s) with congenital teeth agenesis has been excluded22. Tooth Development In mammals, tooth development is a complex process with reciprocal interactions between the dental epithelium and mesenchyme involving the shifting of the odontogenic potential between these tissues (figure III). The first sign of tooth development is the appearance of the primary epithelial band within which the odontogenic process initiates with the formation of an epithelial bud. Mesechymal cells then differentiate around the bud to form the dental papilla, the precursor of the tooth pulp and dentin-secreting odontoblasts that appear after a few additional soft-tissue phases that include the cap stage leading to the bell stage when the enamel-depositing ameloblasts are formed. The dentinal matrix then forms at the periphery of the dental papilla during dentinogenesis and subsequently enamel deposition, or amelogenesis, occurs at the dentino-enamel junction after a few micrometers of dentin has been deposited. Finally, apposition of dentin and enamel gives way to tooth eruption and function. Transcription factors and signaling molecules, which operate both intra- and extra-cellularly, are expressed in a spatially-and temporally-restricted pattern in the epithelium and mesenchyme tissues throughout the odontogenic process and guide tooth development (figure III). Tissue-recombination experiments have helped greatly in developing our understanding of the hierarchy and roles of the various factors with the odontogenic process23-24. BMP4, a member of the transforming growth factor-β (TGF-β) family, and the transcription factors PAX9 and MSX1, members of the paired-box domain and homeobox domain gene families respectively, are examples of controlling factors during the odontogenic process. The odontogenic potential shifts from the epithelium to the dental mesenchyme missing tooth is reported to be the maxillary lateral incisor concomitantly with BMP4 expression25. Both PAX9 & MSX1 by some investigators30,35-36 or the mandibular second are expressed in the dental mesenchyme and their expression premolar by others37-38. The lowest incidence of tooth is key to maintaining the odontogenic potential following agenesis occurs in the lower central and lateral permanent this shift25. PAX9 has been identified as a key controlling incisors with agenesis of maxillary permanent central incisors, factor during the odontogenic process with its expression maxillary permanent cuspids and maxillary permanent first found specifically at the prospective sites of all teeth prior molars also rare39. to there being any morphological signs of odontogenesis25. A general role for MSX1 in the development of ectodermal derivatives has been suggested14 with it strongly expressed in the dental mesenchyme but notably absent from the dental epithelia during the bud, cap and bell stages of tooth development26. Tooth development in both PAX9- and MSX1- mutant mice is arrested at the bud stage27-28, suggesting they have similar, non-redundant roles in signal progression to the cap stage of tooth development. Interestingly, PAX9 and MSX1 have been reported to have an important regulatory role in the maintenance of BMP4 expression and signaling25 implying they may also have a role in odontogenic potential shifts. Clinical features of Hypodontia A congenital anomaly affecting the formation of the dentition that results in a reduction in the usual number of the human permanent dentition (a total of 32 teeth in both jaws) and/or the deciduous dentition (20 total teeth in both jaws) is commonly referred to as hypodontia. Recent studies have shown that the occurrence of hypodontia has increased during the 20th century29. 80-85% of hypodontia cases studied involved the agenesis of just one or two teeth30, indicating that most people afflicted suffer from a mild form of the disease. The tooth most commonly missing is the third molar (or wisdom tooth) which is absent in as much as 20% of the population2,31-34. The second most commonly missing tooth is reported to be the maxillary lateral incisor by some investigators30,35-36 or the mandibular second premolar by others37-38. The lowest incidence of tooth agenesis occurs in the lower central and lateral permanent incisors with agenesis of maxillary permanent central incisors, maxillary permanent cuspids and maxillary permanent first molars also rare39. Hypodontia affecting the primary dentition is rare (prevalence rate of <0.5%) and has been observed to afflict both sexes equally39-40. It is often followed by hypodontia in the same region of the permanent dentition, which itself has a prevalence, excluding third molars (wisdom teeth), ranging between 2.3% and 10%41 and a third of sufferers typically have at least one first-degree relative also afflicted40,42. Severe hypodontia, also referred to as oligodontia, involves the agenesis of six or more teeth and like hypodontia of the primary dentition it is rare afflicting approximately 0.5% of the population43. Hypodontia has been identified as both non-syndromic, where it is an independent congenital oral trait, or syndromic, where it is acquired as part of a specific disease. It is an associated finding in at least 49 syndromes listed in the Online Mendelian Inheritance in Man database18 implying some factors involved in tooth development have a wider role within the human body. Other anomalies associated with hypodontia include small tooth size (microdontia), large tooth size (macrodontia) and anomalies in tooth shape, most commonly tapering or “peg-shaped” teeth40,44. The non-syndromic form of hypodontia can be sporadic or familial and it has been most frequently reported as inherited in an autosomal dominant45-52 (AD) fashion where it displays phenotypic heterogeneity as measured by the nature of the missing teeth and other alterations in the teeth. However, autosomal recessive53 (AR), X-linked54-56 and polygenic57-60inheritance has also been reported. Geographical, population and gender prevalence Variation is seen in the number of teeth found in both the primary and permanent dentition, although it is less common in the primary dentition39, 41. Several population studies have been carried out in the past to establish the epidemiological, clinical and genetic characteristics and prevalence of hypodontia both in primary and permanent dentition through the collection of the dental history of individuals belonging to several families representing various countries/ geographical locations and races. Table I shows the summarized data representing the prevalence of hypodontia in primary and permanent dentition in various geographical locations. It is apparent from these studies that the population prevalence of hypodontia varies with geographical location. In the primary dentition, Japan shows the greatest prevalence of hypodontia which is almost three times greater than the next highest, Finland. Great Britain shows the lowest prevalence which is an eighth of that of Japan with the remaining countries in table I also showing low prevalences between 0.4-0.6%. In the permanent dentition, Saudi Arabia shows the lowest prevalence whilst Iceland exhibits the highest. The greatest differences are found in the populations of both Iceland and Sweden which exhibit a prevalence of hypodontia in their permanent dentition that is 16 or 19 (respectively) times greater than in their primary dentition. Europe has a prevalence of hypodontia in the permanent dentition that is at least ten times greater than any of the European component countries have exhibited in the primary dentition. This pattern is also seen between the primary dentition of New Zealand and the permanent dentition of its neighbor country Australia, whereas Japan has a prevalence in its primary dentition that is only a third of that in the permanent dentition of its neighbor country China. Interestingly, Saudi Arabia exhibits an equal prevalence of hypodontia in both its primary and permanent dentition. It therefore appears that people of Scandinavian decent are the most susceptible to hypodontia in the permanent dentition whilst those of Asian or Arabic descent are the most susceptible in the primary dentition. There are apparently no published reports of large scale studies of populations in Latin American countries. A number of investigations have attempted to take into account any possible gender preference in tooth agenesis (table II). Several reports mentioned a lower prevalence of tooth agenesis in males30,38,61 with one study reporting a male to female ratio62 of 2:3 whilst others have failed to confirm this63. The data in (table II) shows no clear pattern of gender preference with fluctuations between male and female bias where most studies show ratios that are close to parity. Oligodontia The congenital lack of more than six permanent teeth (oligodontia; a severe hypodontia) is also quite prevalent in the population. Studies on different populations have shown variation in the prevalence of oligodontia with the difference in the frequency of oligodontia between males and females found to be not statistically significant64-65, nor is the difference in distribution of missing teeth over mandible/ maxilla and right/left sides64-65. Collective data from six different studies does however indicate that the frequency of oligodontia is lower in males than females and that the frequency of missing second premolars or upper lateral incisors is higher in congenital oligodontia66. Etiology Both genetic and environmental factors have been found to contribute to the etiology of tooth agenesis with many theories having been suggested to explain their affects, particularly prior to the intensive genetic studies performed in recent years38,49,67-68 Environmental Factors Environmental factors can cause tooth agenesis by a variety of means69 that can be broadly placed into two groups: invasive and non-invasive. These can act either additively or independently to affect the positioning and physical development of the tooth. Jaw fractures, surgical procedures, extraction of the preceding primary tooth and changes in muscle pressure from the facial and lingual sides are all examples of invasive factors that can affect tooth development and positioning leading to tooth agenesis and impaction38,49,65. It has also been shown that developing teeth are irreversibly affected by chemotherapy and irradiation in an age- and dose-dependent manner, with the latter having been shown to cause the more severe effects4-5. Thalidomide (Nphthaloylglutamimide) has been reported to cause congenitally missing teeth in children whose mothers took it during their pregnancy1,70-71. Nutrient deprivation and serious illness have also been linked to tooth developmental problems, although no definite etiological relationship has been found between hypodontia and systemic diseases38,49, endocrine disturbances72 or ectodermal dysplasia73. A developmental relationship has been proposed between nerve and hard tissues74 with tooth agenesis linked to the development of the main innervation paths75-76 where it was noted that the regions most commonly affected by hypodontia were the last to undergo innervation. Brainstem anomalies have been shown not to affect tooth development77 indicating that it is local rather than global nerve development that affects tooth agenesis. Genetic Factors In the majority of cases, hypodontia has a genetic basis. Tooth agenesis is found more commonly among individuals related to hypodontia patients than in the population in general56 identifying it as a genetic disease. An exhaustive study on a Swedish family with 685 family members, including 171 probands affected with hypodontia, showed that hypodontia involving permanent teeth is primarily determined by genetic factor(s)38. The frequency of hypodontia among races varies30,78 and greater concordance of hypodontia is apparent in identical twins than nonidentical79-80 with no environmental etiology apparent in afflicted individuals. Familial hypodontia is reported to exhibit mainly autosomal dominant inheritance with incomplete penetrance and variable expressivity49-52. However, an autosomal recessive mode of inheritance for hypodontia has been reported in a Pakistani family which mapped to chromosome 16q12.153 and in another report on Finnish patients that are afflicted with a specific type of hypodontia (Recessive Incisor Hypodontia; RIH), where patients notably lacked both deciduous and permanent incisors81. It has also been suggested that it can follow sex-linked42,59,82 (Patel et al., unpublished results) or polygenic inheritance patterns54-55,57-58. Recently, direct evidence was gathered for the genetic basis of tooth agenesis thanks to the mapping of human disease genes using linkage analysis followed by mutation analysis of positional candidate genes present in the candidate interval. Using this gene mapping strategy, autosomal dominant hypodontia has been localized to at least three chromosomal loci to date; MSX183, PAX913 and an unknown locus on chromosome 106. Five mutations have thus far been identified within MSX1 and ten within PAX9 (table III) with both genes also having been found to be deleted in separate studies of familial hypodontia45,84. MSX1 Although one report has excluded the MSX1 gene as the gene responsible for tooth agenesis85, recent research has identified MSX1 as the causative gene for some forms of congenital teeth agenesis with five mutations having been identified within MSX1 thus far (table III). All of these mutations have been point mutations with two leading to a substitution mutation within the protein and the remaining three form a stop codon that prematurely truncates the protein. Two mutations fall within the N-terminal region prior to the central homeodomain (M61K & S105X) with the remaining three (Q187X, R196P & S202X) all falling within the homeodomain itself (figure 4(A)). Of the two substitution mutations, the M61K mutation15 falls outside of the homeodomain of MSX1 and how it affects its function remains unknown but it has been proposed that it may be through the disruption of protein interactions15. The R196P mutation83 falls within helix-I of the MSX1 homeodomain disrupting its stability and functional activity86. Of the three premature termination mutations, S105X is the only mutation to occur prior to the homeodomain of MSX1 (figure 4(A)). It was identified in a Dutch family suffering from cleft lip-palate and hypodontia that were found to be heterozygous for the 314C>A nucleotide substitution, which creates a stop codon in MSX1 exon 1 truncating the protein prior to the homeodomain87. The remaining two termination mutations fall within the central region of the MSX1 homeodomain. A 559C>T nucleotide substitution was identified in a Flemish family suffering from cleft lip-palate and hypodontia where it forms a stop codon that truncates the protein within helix-I of the MSX1 homeodomain (N187X; figure 4(A))88. The S202X mutation, caused by a 605C>A nucleotide substitution, was identified in a patient with Witkop syndrome who was suffering from hypodontia89 where it generates a stop codon in helix-I of the homeodomain region (figure 4(A)) truncating the protein and disrupting its functional activity86,90. Interestingly, two of the premature termination mutations were identified in patients afflicted with both oligodontia and cleft lip-palate (S105X & Q187X) with the third mutation having been identified in an individual with Witkop tooth-nail syndrome (S202X). They all suffer from additional pathologies other than hypodontia which also afflicts those with substitution mutations indicating that that whilst the substitution mutations disrupt MSX1 activity they do not abolish it whereas the truncation mutations appear to abolish MSX1 activity leading to a more severe phenotype. This is supported by another study that identified haploinsufficiency of MSX1 as the cause of severe oligodontia within unrelated Finnish patients who also had Wolf-Hirschhorn syndrome84. However, there is no clear correlation between the severity of the hypodontia and the severity of the effect on the MSX1 protein caused by the identified missense mutations. PAX9 In contrast to MSX1, both missense and frame-shift mutations in PAX9 have been associated with hypodontia (table III). Of the seven missense mutations identified to date, one is a premature termination mutation (K114X) and the remaining six are all residue substitution mutations. Of these latter mutations, only five generate a substitution in the protein (L21P, R26W, R28P, G51S & K91E) with one believed to prevent PAX9 expression (1A>G). Three frame-shift mutations have been identified, two of which are caused by the insertion of a single nucleotide (G73fsX316 & V265fsX316) and the other by the deletion of 8 nucleotides with the insertion of 288 foreign nucleotides (R59fsX177). All but one of these mutations (V265fsX316) falls within the N-terminal paired-domain (Figure 4(B)) with the exception falling in the approximate middle of the C-terminal region. This single nucleotide insertion falls within exon 4 of the PAX9 gene creating a frame-shift at amino acid 264 (Figure 4(B)) which leads to premature truncation of the protein91. The other single nucleotide insertion was a guanine nucleotide that extends a series of five guanines to six causing a frame-shift between the N- and C-terminal DNA binding domains of the PAX9 paired-domain (Figure 4(B)) abolishing the C-terminal DNA binding domain13. By far the most severe frame-shift mutation is caused by a 288bp insertion in the paired domain of PAX9 (Figure 4(B)) in place of eight deleted nucleotides which leads to a frame-shift that disrupts the C-terminal DNA binding region of the paireddomain47. Two nucleotide substitutions within the PAX9 paired domain were also identified during this study (L21P and K91E), both of which fall within the DNA binding regions of the PAX9 paired domain (Figure 4(B)), N- and C-terminal respectively, which may affect PAX9’s ability to associate with DNA and thus its transcriptional activity. The only substitution mutation to cause premature termination was an A340T switch that creates a stop codon at lysine 114 producing a truncated PAX9 protein that terminates at the end of the N-terminal DNA binding region of the PAX9 Paired-box domain92 (figure 4(B)). The remaining three missense mutations that lead to a residue substitution in the PAX9 protein were all identified recently. An R26W mutation was identified in the N-terminal DNA binding region of the PAX9 paired domain which has been hypothesized to affect its target DNA specificity93. A mutation two residues further into the N-terminal DNA binding region was identified (R28P) and shown to dramatically reduce the DNA binding ability of PAX994. The final missense mutation (G51S) lies within the boundary region between the N- and C-terminal DNA binding regions of the PAX9 paired domain95. Haploinsufficiency of PAX9 has been reported in two studies although its cause has been by two drastically different mechanisms. One study identified a nucleotide substitution in the first position of the ATG start codon that has been hypothesized to abolish its expression48 whilst another identified a deletion of the entire PAX9 gene45. All but one of the PAX9 mutations appear to disrupt the DNA binding ability of its paired domain, thus reducing its transcriptional activity, which appears to be the likely cause of the hypodontia phenotype associated with them. It is interesting to note that most of the PAX9 frame-shift, deletion and missense termination mutations cause hypodontia in both the permanent and the primary dentition, whereas missense substitution mutations affect the permanent dentition only. Locus 10q11.2 A study on He-Zhao deficiency, a distinct form of permanent teeth agenesis which is different from other previously described disorders, in members of a large Chinese kindred has identified an unknown locus on chromosome 10q11.2 using multipoint linkage analysis6. With the increase in prevalence of hypodontia observed over the 20th century, the identification of its causative factors is essential for providing treatment to those afflicted in the future. Modern molecular genetic techniques have allowed us to start to identify the genetic factors responsible for tooth agenesis but more work is required to discover how malfunctions in these factors disrupt tooth development. The identification of more families afflicted with hypodontia is key to identifying the molecular processes that underlie this pathology and our knowledge of tooth development. More patients/families presenting familial hypodontia need to be referred to the geneticist for investigative research to increase the known population of mutations affecting the dentition. To achieve this, dental professionals need to collaborate with human geneticists like ourselves after the identification of probands presenting familial hypodontia and join in our gene discovery efforts. Acknowledgements Work in the authors’ laboratory was supported by NIH grant DE014102 (to PIP). References
Copyright 2005 - Piracicaba Dental School - UNICAMP São Paulo - Brazil |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}