 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
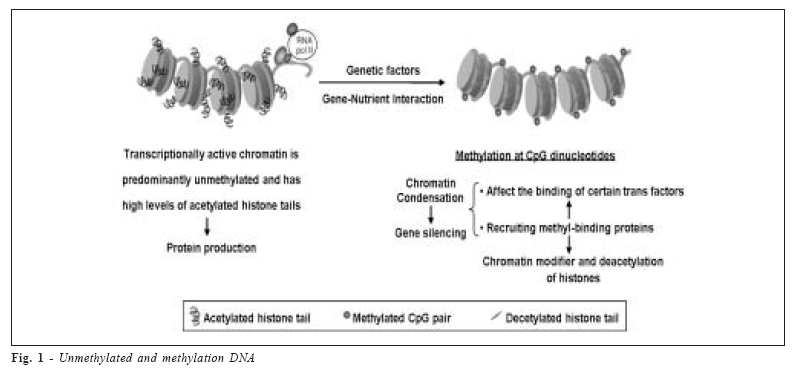
Brazilian Journal of Oral Sciences, Vol. 5, No. 17, Apr-June, 2006, pp. 991-995 The epigenetics of enamel formation Maria Cristina Leme Godoy dos Santos1* Sergio Roberto Peres Line2* 1 Postgraduate Received for publication: October 17, 2005 Accepted: May 16, 2006 Code Number: os06012 Abstract Amelogenins, a family of enamel extracellular matrix proteins, are abundantly expressed during tooth development. It is produced by the AMELX Xq22 and AMELY Yp11 genes and the level of transcription of AMELY locus appears to be only approximately 10% of amelogenin transcripts. DNA methylation is the major epigenetic mechanism of regulation of gene expression. Many factors can interfere in gene silencing, such as polymorphisms and nutrients and some genes escape inactivation, principally X-linked genes. The large number of genes that escape inactivation, and their non-random distribution on the chromosome X may have implications in abnormalities caused by genes on this chromossome. Defects of dental enamel formation are caused by genetic or environmental factors, including genetic polymorphisms and nutrient intake. However, the relationship between epigenetic and genetic factors in amelogenesis is not known. In this context, DNA methylation might be a promising path to explain not only the mechanisms of AMEL inactivation, but also in pathological situations. Key Words: amelogenesis, amelogenin, DNA methylation. Introduction Amelogenin in Enamel Formation Dental enamel, the most highly mineralized structure in the human body, it is formed within a unique extracellular matrix formed by both organic and inorganic components. Its formation is divided into secretory, transition, and maturation stages1 , involving a complex interaction between organic and mineral components. During the secretory stage, there is a predomination of proteins components, which go through selective proteolysis during maturation. This progressive protein hydrolysis build a sketched of protein matrix and permit hidroxyapatite crystals to grow1 . Amelogenins, a family of extracellular matrix proteins of dental enamel, are transiently but abundantly expressed during tooth development2. They are secreted by the ectodermderived ameloblasts in tooth buds during early stages of odontogenesis and are the most abundant enamel proteins - accounting for more than 90% of total enamel protein3 . The profile of enamel matrix proteins changes during tooth formation. Early enamel contains predominantly the prolineric amelogenins, which are selectively metabolized during maturation4 . Amelogenin is initially secreted as a 25-kDa nascent protein, which has a high degree of polarity determined by its highly hydrophilic C terminal teleopeptide. The teleopeptide seems to be processed rapidly by specific endoproteinases and/or carboxy-proteinases to form a 23- kDa amelogenin molecule intermediate5. A 20-kDa amelogenin stable form results from either transient 23-kDa intermediates or a single cleavage of 25-kDa parent amelogenins6. The accumulation of 20-kDa amelogenin in enamel indicates a relative slowdown in further protein hydrolysis7. This fragment is cleaved to produce both an insoluble tyrosine rich amelogenin peptide (TRAP) and a soluble 13-kDa fragment7. Amelogenin is, therefore, degraded extracellularly and enzimatically, resulting in a number of smaller peptides. Amelogenin regulates the formation of enamel crystallites by producing the skeleton necessary for mineralization and crystal formation of the enamel. Amelogenin protein aggregates referred to as nanospheres support and protect growing crystals, inhibiting intercrystallyte fusions, creating channels for ion transport7. Amelogenin is thought to form a scaffold for enamel crystallites and to control their growth6, but its exact functions are not fully known8. Expression of Amelogenin Amelogenin is produced by the AMELX Xq22 and AMELY Yp11 genes. The genes contain seven exons spanning > 9Kb2. Although the transcriptionally active amelogenin gene is found on both human X and Y chromosomes, the level of transcription of AMELY locus appears to be only approximately 10% of amelogenin X2. This suggests that in the developing tooth bud of males, AMELY and AMELX undergo a non-random patter of dosage compensation with preferential inactivation of the AMELY locus9. The other interesting point is that at least 14 mutations have been described in AMELX, resulting in amelogenesis imperfecta, a group of inherited defects of dental enamel formation that show both clinical and genetic heterogeneity10 . However, there are no reports of amelogenesis imperfecta with mutations in the Y amelogenin gene. This may suggest a preferential inactivation of the AMELY and a form of gene regulation similar to that of genomic imprinting, resulting in differential expression of parental alleles during both embryogenesis and adult life9. On the other hand, the mosaic patters of normal and abnormal enamel formation in female carriers of X-linked amelogenesis imperfect evidence X inactivation2. Besides, individuals with one extra X chromosome – 47, XXX or 47,XXY – were found to have thicker enamel than normal males and females, suggesting that the extra X chromosome may be active in amelogenesis11 . The differences in the transcription levels of the AMELX and AMELY genes very likely resides in the promoter regions, which share only 80% sequence similarity. Both promoters contain identical TATA and CCAAT boxes. However, other cis-acting elements, that may control the transcriptional activity of the amelogenin genes, have not been characterized2 . Gene Inactivation The human X and Y chromosomes share significant sequence similarity, likely because of their evolutionary origin from a common ancestral homologous chromosome pair2. Key to their evolution, are structural rearrangements and successively suppressed recombination, allowing independent evolution of each chromosome and leading to the largely degraded Y chromosome12 , while the X chromosome has been heavily influenced by X inactivation. In female mammals, most genes on one X chromosome are silenced as a result of X-chromosome inactivation13 . However, about 15% of X-linked genes escape inactivation in some degree (partial and incomplete) and are expressed from both the active and inactive X chromosome. The proportion of genes escaping inactivation differs dramatically between different regions of the X chromosome. An additional 10% of Xlinked genes show variable patters of inactivation and are expressed to different extents from some inactive X chromosome. These data indicate that females have considerable heterogeneity in levels of X-linked gene expression14. An important factor that can influence inactivation status is the presence of DNA methylation. DNA methylation is a major epigenetic mechanism of regulation of gene expression that has gained increasing importance in differentiation processes15. In vertebrates DNA methylation occurs in cytosines, most often when a cytosine is followed by a guanosine (CpG dinucleotide). Approximately one-half of human genes have CpG islands in their 5’-promoter regions or within the first exons16. CpG islands usually are unmethylated17, and the methylation of these CpG-rich sequences induces inhibition of their expression. The patterns of DNA methylation, therefore, distinguish at a molecular level the genes to be expressed selectively18. Alterations in DNA methylation have been described as regulating a differentiation path in a particular tissue19. DNA in germ line cells usually is fully methylated, and demethylation usually is observed in a tissue-specific fashion. However, most of the housekeeping genes frequently are maintained in a completely unmethylated state in both the germ line and in issue-specific sites20. Mechanistically, there are a number of ways in which DNA methylation can repress transcription. CpG methylation within gene promoters can act directly by affecting the binding of certain transcription factors to DNA or by recruiting methyl-binding proteins that in turn attach to chromatin modifier complexes including histone deacetylases, causing deacetylation of adjacent histones and subsequent chromatin condensation and gene silencing21. Many of the trans factors known to bind to sequences containing CpG dinucleotides do not bind when the CpG doublets are methylated22. Alternatively, methyl-CpG-binding proteins, such as MeCP2, bind preferentially to methylated DNA and directly repress transcription, inhibit the binding of others trans factors, structurally modify the DNA, or recruit corepressor complexes23-25. DNA methylation is involved in major physiological processes including X-chromosome inactivation, genetic imprinting and tissue-specific gene expression26. Therefore, DNA methylation is a fundamental mechanism for the epigenetic control of gene expression and the maintenance of genomic integrity27-28. Recent finding support the premise that hypomethylation of the DNA surrounding the proximal promoter region is a prerequisite for gene activation, whereas heavy methylation leads to gene silencing29. Besides, histones modifications represent an additional pathway of epigenetic regulation of gene transcription30. Intriguingly, the lysine residues on histones can be acetylated and methylated; H3 lysine 4 methylation has been correlated with active expression31, whereas H3 lysine 9 methylation has been linked to gene silencing and the assembly of heterochromatin32. It is now believed that DNA methylation pathways and the histone code are functionally interactive33. The mechanism for the CpG island-associated gene silencing seems to involve the link of specific methylated DNA binding proteins, followed by the recruitment of a silencing complex that includes histones deacetylases34-35. The de novo methylation, by itself, has a minimal effect on gene expression. However, methylated DNA recruits methyl-binding proteins, which also attracts a protein complex that contains histones deacetylases. Through the action of methyl-binding proteins and histones deacetylases, the DNA structure changes to a compact, condensed chromatin configuration that results in permanent inhibition of mRNA and protein production36. The local cytosine methylation of a particular sequence can directly interfere with the binding of certain transcription factors37, so hypermethylation of the coding region can decrease gene transcription. Conversely, hypomethylation of the coding region also can increase the gene transcription by enhancing the binding of transcription factors19. Hypomethylation of the coding regions of critical genes can lead to instability either because this region becomes more susceptible to endogenous nucleases38 or because the site of hypomethylation is likely to undergo enzymatic deamination to uracil39-40. Couwenhven et al.41 observed that amelogenin transcriptional activity in vivo and in vitro did not correlate with changes in methylation of the amelogenin gene. However, more studies are needed to determine the effect of DNA methylation in amelogenesis. Factors Associated DNA Studies have shown that DNA methylation can be associated with specific alleles. Stern et al.42 showed that mutation in methylenetetrahydroolate reductase (MTHFR) is associated with folate status and affect methylation of DNA. MTHFR catalyzes the synthesis of 5-methyl THF, the methyl donor for methionine synthesis and precursor of Sadenosylmethionine, necessary to DNA methylation. This gene is reported to have 2 polymorphisms – C677T and A1298C – that lead to reduce MTHFR activity43-44. It was observed that subjects homozygous for the MTHFR C677T and A1298C polymorphism possessed a lower degree of genomic DNA methylation compared with the CC and AA wild-type person, respectively45. Methionine synthase catalyzes the transfer of methyl base from 5-methyl THF to homocysteine. Its gene has a polymorphism in 2756 A to G, resulting on a lower enzyme activity46, and this result in homocysteine elevation and DNA hypomethylation. Since these polymorphisms are associated with diverse pathologic conditions, these data provide evidence of the existence of a putative inherited differential susceptibility to DNA methylation between healthy and diseased individuals. Gene-Nutrient Interactions and DNA Methylation Nutrition research has recently highlighted the role of several nutrients in regulating the genome machinery. A number of vitamins and micronutrients are substrates and cofactors in the metabolic pathways that regulate DNA synthesis and/or repair and expression of gene47. It has been documented that the nutricional deficiency may result in the disruption of genomic integrity and alteration of DNA methylation, thus linking nutrition with modulation of gene expression19. Many micronutrients and vitamins are indispensable in DNA metabolic pathways47-48 and can interfere in gene silencing. Studies in rat showed that zinc deficiency can reduce the utilization of methyl groups from S-adenosymethionine and results in genomic DNA hypomethylation as well as histone hypomethylation49-50; and that dietary deficiency in selenium decrease genomic DNA methylation51-52. Vitamin C deficiency has been associated with DNA hypermethylation in lung cancer cell53-54. Interestingly, niacin, precursor of NAD+, is required to maintain the unmethylated state of CpG dinucleotides by inhibiting the enzymatic DNA methylation. This is necessary for the synthesis of poly-ADP-ribose polymerase-1, which converts histone H1 to poly-ADPrybosylated forms that is responsible for the enzymatic inhibition of DNA methylation54-55 . On the other hand, folate and/or methyl group dietary supply provides the most compelling data for the interaction of nutrients and DNA methylation, because these dietary elements are directly involved in DNA methylation via onecarbon metabolism. The sole metabolic function of all coenzymatic forms of folate is to transfer one-carbon units19. Folate deficiency may increase the risk of malignancy by causing DNA hypomethylation, inducing uracil misincorporation during DNA synthesis, or both, resulting in deficiency in the DNA repair process and, therefore, DNA strand breaks56-57 . It is interesting that there is an increase in methyltransferase activity and methylation of CpG sites after extended periods of folate deficiency58. Alcohol consumption in combination with poor folate intake causes megaloblastic anemia; decreasing intestinal absorption of folate; inhibiting enzymes, such as DNA methyltransferase or methionine synthetase; and trapping folate59. It has been shown that moderate alcohol intake also may diminish serum vitamin B12 concentrations among healthy, postmenopausal women60. Both vitamin B12 and folate are required for DNA methylation and nucleotide synthesis. Therefore, chronic alcohol consumption may induces hypomethylation in genomic DNA. In conclusion, defects of dental enamel formation may be caused by genetic or environmental factors, including genetic polymorphisms and nutrient intake. However, the relationship between nutrient consumption and genetic factors in amelogenesis is not completely understood. The large number of genes that escape inactivation, and their non-random distribution on the chromosome has implications in X chromosome abnormalities. More studies are need to determined whether or not the inactivation of amelogenin X and Y occur in a random patter, analogous to others X-linked genes, which undergo dosage compensation in females. In this context, DNA methylation might be a promising path to explain not only the physiological mechanisms of amelogenin inactivation, but also pathological situations. Acknowledgments This work was supported by FAPESP grant 03/09128-8. (Figure 1) References
Copyright 2006 - Piracicaba Dental School - UNICAMP São Paulo - Brazil The following images related to this document are available:Photo images[os06012f1.jpg] |
| |||||||||

{kind=link}