 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
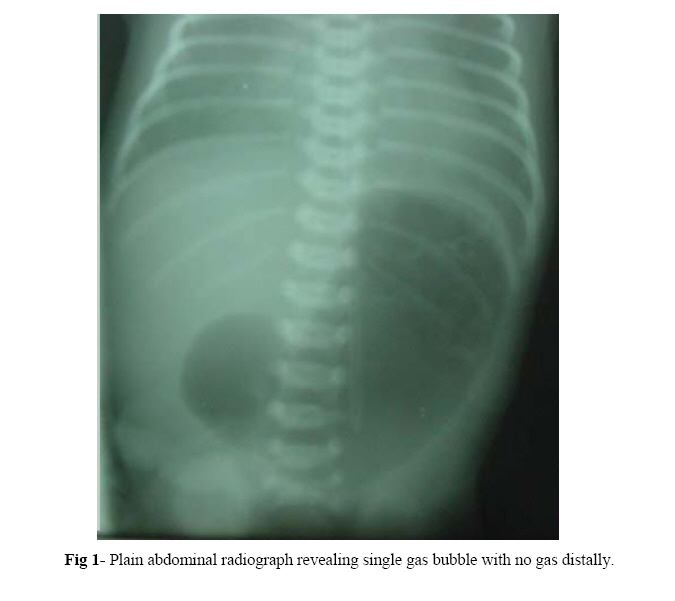
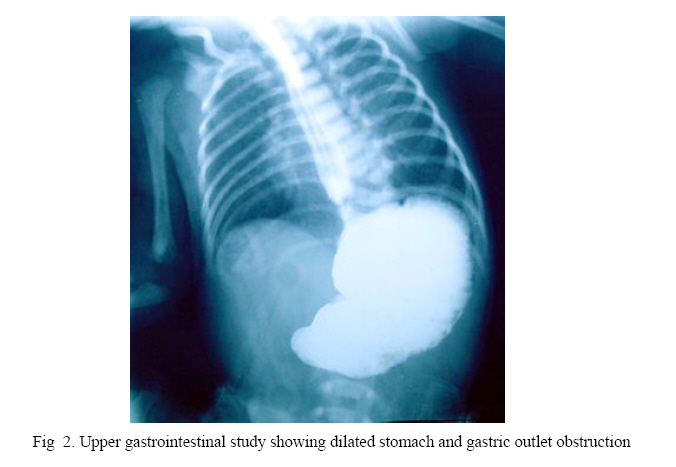
Iranian Journal of Pediatrics, Vol. 17, No.4, December 2007, pp. 369-374 Pyloric Atresia Associated with Epidermolysis Bullosa: A Report of 4 Survivals in 5 Cases Alireza Alam-Sahebpoor*1, MD; Vajihe Ghaffari2, MD; Leila Shokoohi3, MD
* Correspondence author; Address: Department of Pediateric Surgery, Booali Sina Hospital, Sari, Iran. P.O. Box: 4715838477 E-mail: alireza_alam@yahoo.com Code Number: pe07069 Abstract Objective: Pyloric
atresia
(PA)
is
a
rare
congenital
anomaly
that
constitutes
approximately
1%
of
all
intestinal
atresias,
and
its
incidence
is
approximately
1
in
100,000
live
births.
PA
may
occur
as
an
isolated
condition
or
associated
with
other
abnormalities,
the
most
common
being
Junctional
epidermolysis
bullosa
(EB).
Evidence
suggests
that
PA-EB
is
a
distinct
entity.
In
this
report,
we
present
5
cases
of
pyloric
atresia
associated
with
Junctional
epidermolysis
bullosa,
4
of
whom
survived
after
surgery. Key Words: Pyloric atresia, Epidermolysis bullosa, Gastroduodenostomy, Surgery IntroductionPyloric atresia (PA) is a rare malformation which is estimated to be responsible for less than 1% of gastrointestinal atresias with an incidence of 1 in 100,000 live births.[1] Anatomically, PA is divided into 3 types pyloric membrane or web (57%), pyloric canal replaced by a solid cord of tissue (34%), PA with a gap or gap aplasia (9%).[2] Epidermolysis bullosa (EB) consists of a group of hereditary skin disorders, where the primary cause is formation of blisters following minor trauma. EB is divided into 3 main types according to the histopathologic location of the bullae: Epidermolysis bullosa simplex (EBS), Junctional epidermolysis bullosa (JEB), and Dystrophic epidermolysis bullosa (DEB). Unlike JEB as an autosomal recessive trait, EBS and DEB can be inherited either as autosomal recessive or as autosomal dominant. All three types have been reported in association with PA, although JEB is most frequently reported[3]. Evidence suggests that PA-EB association is a distinct entity and is now referred to as the PA-EB syndrome[4]. Because of septicemia, electrolyte imbalance and protein loss seen in EB, the association of PA-EB was thought to be fetal and it was recommended in the past that surgical treatment be withheld in these patients. Recently there has been encouraging reports of survival among these patients. Case(s) PresentationThe study was conducted in the pediatric surgery units of Boo-Ali hospital in Sari/Iran from 2003 to 2005, as an interventional study. Five patients with PA associated with JEB were referred to us from different hospitals. Case 1: A premature baby girl was born by Caesarean section at 32 weeks of gestational age. The pregnancy was complicated by severe vaginal bleeding. Antenatal sonography was normal and there was no polyhydramnios. The neonate had copious, nonbilious vomiting since birth with mild epigastric dilation. She was referred to us for surgical consultation on 8th day of life. Physical examination showed multiple bullous eruptions over all extremities. Biopsy from one of these lesions was compatible with JEB. An abdominal X-ray showed distended stomach with no distal gas (Fig 1). Laparatomy revealed PA type 2 and gastroduodenostomy was performed. Oral feeding started on the 7th postoperative day and was well tolerated. Skin lesions healed without scar. She was seen regularly in outpatient clinic and now at 3 years of age is doing well. Case 2: This 15-day old boy was referred to us because of nonbilious vomiting. Since the 8th day of life he developed bullous skin lesions all over the body especially over the ankles and hands. He was born by normal vaginal delivery (NVD), with a birth weight of 2000 grams. Antenatal ultrasound (U.S) was normal. Mother had urinary tract infection during pregnancy and received antibiotics. Abdominal plain X-ray showed single bubble of gas. In the referring hospital an upper GI series had revealed dilation of stomach with no contrast beyond the pylorus (Fig 2). Biopsy of a fresh bulla showed JEB. At laparatomy there was a dilated stomach with a membrane at the pylorus (type 1). The pyloric membrane was excised and Heineke-Mikulicz pyloroplasty performed. Oral feeding started on the 8th postoperative day and was well tolerated. The patient was discharged 14 days after operation. On follow-up 1 month and 3 months post operation he was doing well and all his skin lesions were healed. He tolerated formula with no vomiting. Case 3: A 3-day old boy was referred to our hospital for surgical consultation because of nonbilious projectile vomiting. He was born by C/S at 36 weeks gestational age. He weighed 2700 grams. Antenatal ultrasound scans showed polyhydramnios. A plain abdominal X-ray revealing single bubble with no distal gas confirmed the diagnosis. Laparatomy was done at day 5 of life. A type 2 PA was noted. Gastro-duodenostomy was performed. The next day he showed bullous eruptions on the palm and on pressure sites as well. Biopsy of one of the lesions was compatible with JEB. On day 5 post operation we started oral feeding. The child had an uneventful postoperative course and was discharged. On follow-up 6 months after operation he is doing well and has gained weight. Case 4: A baby boy was born by NVD at term. His weight was 2 kg. At the second day of life he had copious nonbilious vomiting and on 11th day he was referred to us. No antenatal ultrasound was done. He also had bullae on his hand, feet and trunk. Biopsy of one fresh bulla showed JEB. Because of a distended stomach as a single bubble on plain radiography we suspected PA. Laparatomy confirmed type 2 PA and gastroduodenostomy was performed. On day 5 post operation oral feeding was started. He was discharged home and on follow-up 2 weeks later was doing well. He has regular visits to clinic and 6 month post operation has no problem. Case 5: This 5-day old boy was born by NVD, and referred to us for nonbilious vomiting. Antenatal ultrasonography showed poly-hydraminios. On Physical examination there were multiple bullous eruptions over the extremities. Biopsy of one of the lesions was compatible with JEB. Abdominal X-ray showed single bubble of gas. At laparatomy there was a type 2 PA. Gastroduodenostomy was performed with tube gastrostomy. One week post operation he became septic and died of fulminant septicemia on 12th postoperative day. They were 4 boys and one girl. Mean age of presentation was 8.4 days (3 to 15 days). Prenatal ultrasound showed polyhydramnios in 3 and was normal in 2 cases. There were multiple bullous skin lesions over the extremities in all of the patients. Biopsy of the fresh bullae was compatible with JEB in all 5 patients. All patients underwent laparatomy after stabilization. Four neonates with type 2 PA underwent gastroduodenostomy, and the one patient with type 1 PA had excision of membrane and pyloroplasty. By 6 to 36 months follow-up (Mean 21.6 months) all 4 infants survived and were doing well. DiscussionThe earliest description of PA is credited to Caleder in 1733[5], and Swinbure and Kohler first described the association of PA and EB in 1968[6]. This association was then extensively described by Teran.[7] The typical presentation of PA is non bilious vomiting soon after birth without abdominal distention. The diagnosis is made easily on plain abdominal X-ray, which shows a single gas bubble representing the distended stomach with no distal gas. Upper GI series is almost never indicated as further investigation. It does not provide additional information because air is an effective and safe contrast medium. Antenatal ultrasound may show polyhydramnios and dilated stomach.[8] Prematurity and poly-hydramnios are found in most of these patients like what we saw in our first and third patients. EB is an autosomal recessive gene affecting the integrity of the basement membrane and hemidesmosomes and the control of the normal process of fibrosis occurring during the course of wound healing. Diagnosis of EB is based on clinical and laboratory criteria. The lesions constitute of blisters, crusted erosions and scar formation. The conventional histological techniques used for the diagnosis of EB are transmission electro-microscopy and immunofluorescence mapping. Of the 3 types of EB, JEB is mostly associated with PA. Defects in at least six distinct genes have been identified in JEB. All code for proteins involved in the hemidesmosome/ anchoring filament complex.[9, 10] Prenatal diagnosis is possible for PA-EB first by fetal DNA analysis and second by analysis of fetal skin biopsy using electromicroscopy and indirect immunoflueresence. Major ultrasound sign of PA is gastric dilatation. In 1990 Meizner and Carine described the Snow Flake sign, an echogenic appearance of the amniotic fluid during the second trimester of pregnancy associated with EB.[11] Second trimester amniotic fluid is usually anechoic and visualization of any particle in it is pathologic. Sicot et al. emphasize the difficulty of inter-preting prenatal ultrasound findings where there is no suggestive context.[12] In the literature most of the prenatal diagnoses reported occurred in cases with a positive family history of PA-EB. The poor prognosis associated with this syndrome has led some to attempt to diagnose this condition prenatally when there is a positive family history. PA-EB is known to be caused by the ITGA6 or ITGB4 genes encoding for the integrin alpha/6 or beta/4 subunits, respectively.[13, 14] There are many theories regarding the cause of PA in patients with PA-EB. One hypothesis suggests that PA occurs as a result of intrauterine complication of EB where the pyloric mucous membrane is affected leading to sloughing with subsequent scarring and fibrosis and obliteration of the pyloric canal.[1, 15] The surgical treatment of PA depends on the type of atresia. In type 1 PA, membrane is excised and Heieke-Mikulicz pyloroplasty performed.[16, 17] For type 2 atresia the surgical treatment is excision of atretic segment and gastroduodenostomy.[16,18] For type 3 also the surgical treatment is gastroduodenostomy.[18] Gastrojejunostomy is better to be avoided because it is associated with 59% failure and 55% mortality.[19] The association of PA with EB is well documented and a plain abdominal X-ray should be performed in all babies with EB to exclude PA. The distribution of blisters in EB may be localized or generalized. Blisters are frequently present at birth, but it is important to remember that they may manifest up to 48 hours after birth.[2] Although the JEB with its autosomal recessive pattern of inheritance has been reported widely in association with PA , other types of EB have also been documented.[3] Type 1 atresia predominates in non-EB cases, and type 2 cases are mostly associated with EB. Four of our patients had type 2 PA and only our second patient had type 1 PA. Whereas the surgical management for PA is straightforward, septicemia, electrolyte imbalance, protein loss and failure to thrive complicates the severe exudative skin lesions, often leading to death, and it has led to recommendation that surgical treatment be withheld in patients with PA-EB. Four of our cases were stable before operation, and tolerated it well. They were all well on follow-up visits. Follow up was from 6 to 36 months (mean 21.6 months). Our fifth case was not in good condition before operation and died of sepsis after operation. Postoperatively all 4 survived patients tolerated oral feeding well and went home with no problem. Hayashi et al[20] and other investigators also recommend that stable infants with PA-EB should be treated surgically. Smad et al reported 3 cases of PA-EB. One of them died before operation, and one of the two that had operation survived .They concluded that although the prognosis is poor in this condition, surgery should not be withheld on assumption of high mortality.[4] In some cases of PA-BA, a third phenomenon is described: Alasia cutis congenita (ACC). Maman et al. reported 5 newborns and 3 fetuses with EB-PA-ACC association.[22] A variety of urinary tract diseases like ureterovesical junction obstruction leading to the early development of hydronephrosis should be suspected. So ultrasonography and cystoscopy are recommended in all survivors with PA-EB association. Wallerstein et al. reported 2 cases of epidermolysis bullosa-pyloric atresia-obstructive uropathy (EB-PA-OU).[21] The syndrome of EB-PA-OU is a rare genetic entity of variable severity manifested by skin finding of multiple bullae and, infrequently, aplasia cutis congenita, the gastrointestinal finding of pyloric or other atresias, and progressive obstructive uropathy. None of our 4 survived patients developed urologic symptoms up to now. If PA, which is a rare condition, is diagnosed and treated on time, mortality and morbidity becomes low. The main cause of death is septicemia and electrolyte imbalance associated with EB. ConclusionWe add these 5 patients to the number of children with EB-associated PA who underwent surgery, survived and now are doing well. We recommend surgical treatment for stabilized neonates with PA-EB. References
Copyright 2007 - TUMS PUBLICATIONS The following images related to this document are available:Photo images[pe07069f1.jpg] [pe07069f2.jpg] |
| |||||||||

{kind=link}
{kind=link}