 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
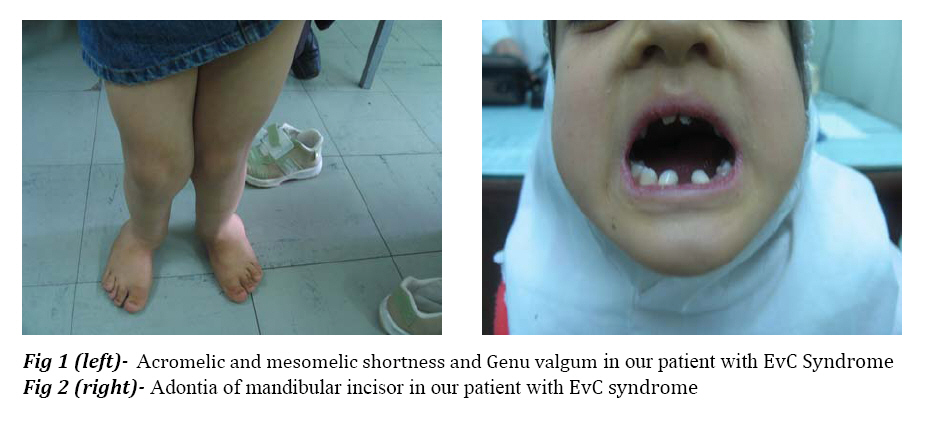
Iranian Journal of Pediatrics, Vol. 18, No. 1, March, 2008, pp. 75-78 Case Report Ellis van Creveld Syndrome: Report of a Case and Brief Literature Review Hedyeh Saneifard*1, MD, Fellowship in Pediatric Endocrinology; Gholamhossein Amirhakimi2, MD, Pediatric Endocrinology and Metabolism 1Shiraz University of Medical Sciences and Health Service, IR Iran Received: 04/11/07; Revised: 27/12/07; Accepted: 20/01/08 Code Number: pe08013 Abstract Objective: Ellis van Creveld syndrome (EvCS)
is a rare autosomal recessive (AR) disorder first described in 1940. The
syndrome manifests with several skeletal, oral mucosal and dental anomalies,
congenital cardiac defects and nail dysplasia. EvCs should be differentiated
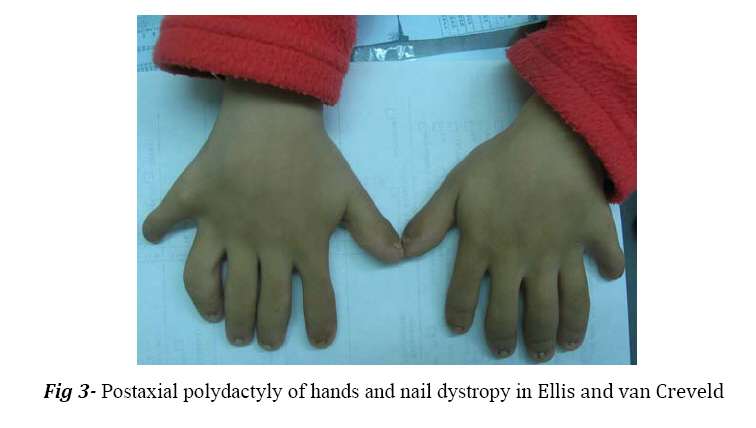
from other chondrodystrophies such as achondroplasia and Morquio's syndrome. Key Words:Postaxial polydactyly, Chondroectodermal dysplasia, Skeletal anomalies Introduction In 1940, Ellis and van Creveld defined a syndrome with certain type of chondrodysplasia, bilateral postaxial polydactyly of hands, small and dysplastic nails and congenital heart defects[1,2]. This syndrome is a rare autosomal recessive disorder caused by mutation in the EVC and EVC2 genes (4 p16)[3]. Affected individuals with mutation in EVC and EVC2 have the typical spectrumof features of this syndrome and are phenotypically indistinguishable[4]. The incidence of EvCS in general population is low[3]. This syndrome is most prevalent in the Amish population of USA. The birth prevalence in non-Amish population is estimated to be 7/1.000.000 of live birth[5,6]. Polydactyly is a constant finding in the hands, and is usually bilateral, postaxial and on the ulnar side. Polydactyly of the feet is present in only 10% of the patients[7]. The extremities are often plump and acromelic; mesomelic shortness of limbs is often encountered. Shortening is most common in distal aspect of the limbs[1]. The nails are hypoplastic, dystrophic, friable, and sometimes completely absent[1,7]. Short stature is due to shortness of lower legs. Shortness is present at birth and becomes more apparent with subsequent growth. Genu valgum, curvature of humerus, and pectus carinatum with a long narrow chest and short poorly developed ribs are present[1,2]. The hair is thin and sparse[8]. Fusion of the middle portion of the upper lip to the maxillary gingival margin or the presence of numerous frenula tethering the upper lip to gingiva are the oral manifesttations of this syndrome[1,8].Hypodontia involving the maxillary and mandibular incisor is a consistent finding[1,2,8]. In the edentulous mandibular incisor region, multiple small alveolar notches may be present on the crest of the thin alveolar ridge giving a serrated appearance[1,5]. Congenital heart defects are reported in about 50% of the cases, either single atrium or a large atrial septal defect is observed[2]. Most patients have normal intelligence. It is almost impossible to radiographically differentiate EvCS from similar chondrodystrophies such as asphyxiating thoracic dystrophy, achondroplasias, chondroplasia punctata and Morquio's syndrome. Differential diagnosis is made with clinical findings such as cardiac anomalies, nail hypoplasias, fusion of upper lip and gingiva, oligodontia and narrow thorax[9,10]. Case Report A nine-year old female was referred with short stature. There was no history of parental consanguinity or affected siblings in the family. She had normal intelligence and was doing well as a 4th grader at school. Her height was 105 cm. The extremities were plump and showed acromelic and mesomelic shortness of the limbs. Genu valgum was seen (Fig 1). Narrow thorax with short poorly developed ribs giving rise to a pigeon breast deformity was noticed. In oral examination, oligodontia of lower incisor was seen. Abnormal crown form was present with hypoplastic developmental enamel with a high caries rate and poor dental hygene. Anterior teeth were conical (Fig 2). Fingernails and toenails were markedly hypoplastic, thin and wrinkled (Fig 1 and Fig 3). Bilateral postaxial polydactyly of hands were present (Fig 3). No cardiac abnormality was reported in examination by the cardiologist. Discussion The most striking features of EvCS reported in the literature include bilateral postaxial polydactyly of the hands, ectodermal dysplasia affecting the nails and teeth and congenital heart abnormalities[11]. Lower limbs are primarily affected and deformed due to mesomelic shortening. It may also be associated with knock-knees or genu valgum, which requires surgical correction. The hands are short and wide exhibiting polydactyly with additional finger next to fifth finger, which is found in 100% of cases[2]. In the present case most of the reported features of this syndrome such as bilateral postaxial polydactyly of hands, hypoplastic nails of hands and feet, genu valgum, short stature, adontia of mandibular incisors were present. Oral manifestations included fusion of middle portion of upper lip to the maxillary gingival margin eliminating the normal mucolateral sulcus. Intraorally, presence of natal and neonatal teeth and congenital absence of teeth particularly in mandibular anterior segment was seen. Tooth eruption was delayed and those erupted were generally malformed or were affected early by caries[8]. In this case, history of delayed eruption of teeth and early involvement with caries and adontia of mandibular incisors was present. Ellis van Creveld syndrome should be differentially diagnosed from two conditions with overlapping features like asphyxiating thoracic dystrophy and short rib polydactyly syndrome. The central feature of asphyxiating thoracic dystrophy is small chest that appears long and narrow. The main distinguishing feature is the absence of hypoplastic nails of hand and feet and asymmetrical presence of polydactyly, but in EvCS symmetrical presence of polydactyly and hypoplastic nails are noted. The other condition, which should be considered due to common features, is short rib polydactyly syndrome. It is characterized by underdeveloped lungs, polydactyly, cleft lip and palate, kidney and intestinal malformations. But in the present case the lungs, kidney and intestines were normal[1]. Prenatal diagnosis can be made with intrauterine growth retardation, skeletal malformations and cardiac defects on ultrasound images. Diagnosis is also possible using chorionic villi or amniotic fluid using linked-microsatellite markers if a previously affected sibling has been identified[9]. One third of these patients die at the early age or at infancy from cardiorespirtory problems and those who survive require multidisciplinary approach for treatment i.e. orthopedic correction of genu valgum, amputation of extra digits, surgical repair of cardiac malformations and dental intervention for high caries risk individuals[1]. Conclusion EvCs is a rare AR disorder, with a high mortality in early life, 1/3 of these patients die ininfancy from cardiac and respiratory problems and those who survive require multidisaplinary approach for treatment. Management during the neonatal period is mostly symptomatic, orthopedic follow up is required to manage the bone deformity; professional dental care should be considered for management of the oral manifestation. Early treatment can prevent the patient from various complications and undue psychological trauma. References
© Copyright 2008 - TUMS PUBLICATIONS The following images related to this document are available:Photo images[pe08013f1-2.jpg] [pe08013f3.jpg] |
| |||||||||

{kind=link}
{kind=link}