 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
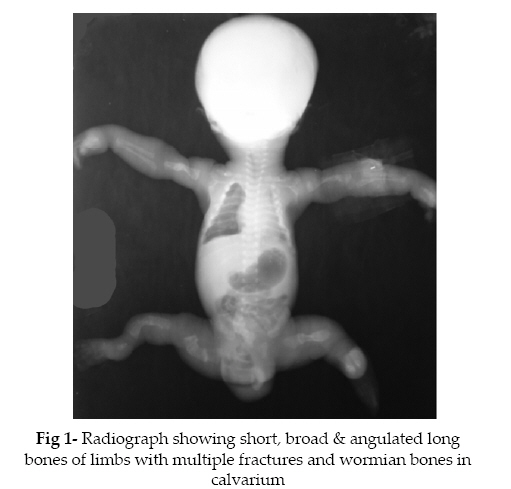
Iranian Journal of Pediatrics, Vol. 18, No. 2, June, 2008, pp. 175-178 Osteogenesis Imperfecta Type II with Congenital Heart Disease Amar Taksande1, MBBS, MD, Pediatrician; Krishna Vilhekar1, MD, Pediatrician; Sona Khangare1, Resident in Pediatrics 1Department of Pediatrics, Mahatma
Gandhi Institute of Medical Sciences, Sevagram, India Received: 05/12/07; Revised: 30/04/08; Accepted: 05/05/08 Code Number: pe08029 Abstract Objective:Osteogenesis imperfect (OI) is an inherited disorder of
type1 collagen synthesis with varied complication. OI type II is a perinatally
lethal variety, characterized by short limbs, broad long bones, radiologic
evidence of severe osseous fragility and defective ossification. These patient
usually are stillborn or die in early infancy of respiratory failure. It has a
wide range of phenotypic expressions, but cardiovascular anomalies tend to be
rare association. When they do occur, they usually consist of aortic or mitral
valve disease. Key Words: Osteogenesis Imperfecta; Atrial Septal Defect; Hernia; Neonate; Fracture Introduction Osteogenesis imperfecta (OI) or “brittle bone disease”, the most common genetic cause of osteoporosis, is a rare disorder of connective tissue involving the bones, ligaments, tendons, skin and sclera. It is characterized by bone fragility and pathological fractures of long bones, blue sclera, thin skin, joint laxity, hernias, wormian bones, and secondary skeletal deformities. The incidence of OI in infancy is about 1 in 20,000[1]. The mild form of OI type I (tarda), is the most common and dominantly inherited while the severe forms of the disease, OI congenita types II – IV, are sporadic, severe and present with fractures at birth[1,2] . Here we report a case of OI type II in a neonate with acyanotic congenital heart disease. Case Presentation A 20 year old primi mother, delivered a 34 week preterm male baby by breech presentation, at Kasturba Hospital, MGIMS, Sevagram. The baby was not cried immediately after birth, bag and mask ventilation given. The baby had multiple congenital anomalies and not cried immediately after birth with an Apgar of 3, 5, 7 and the baby expired after 6 hour of life. Mother was a unbooked patient and received full immunisation during pregnancy. There was no history of consanguinity in the parents and no past or family history of malformation. The antenatal period was uneventful. The baby at birth weighed 1135 gm, and the limbs were short & bowed and the hips were abducted & flexed. On examination, the baby had small nose with low nasal bridge, flat facial profile, bluish sclerae and bilateral proptosis. The respiratory rate was 74/min and heart rate was 156/min, regular with normal heart sounds being heard. No cardiac murmurs were heard. The abdomen and spine were grossly normal. The skull was soft and the fontanelles were large and continuous. Right sided inguinal hernia was present. Echocardiography revealed moderate size (6mm) ostium secundum atrial septal defect without pulmonary hypertension. The infantogram (fig 1) revealed the presence of multiple fractures, involving bilateral humeral shafts, bilateral tibia-fibula in the legs and bilateral femoral shaft. Also, noted wormian bones in the skull. A diagnosis of OI type II with atrial septal defect with right sided inguinal hernia were made. No abnormalities of other internal organs was detected. Because of parent unwilling, autopsy was not done. Discussion OI is one of the inherited disorders of collagen that also includes such diseases as Ehlers-Danlos and Marfan syndrome. It is a heterogeneous group of collagen disorders of different severity characterized by osteopenia and bone fragility, blue sclerae, dentinagenesis imperfecta or opalescent and brittle teeth, progressive sensorineural deafness, and laxity of the joints. There are four types with subdivisions of the types. Each subtype can be divided due to locus and allelic heterogeneity. Type II is the most severe type with certain stillbirth or neonatal death[1]. Clinical picture, radiological evidence and positive family history form the basis of diagnosis and classification. Sillence classification is the commonest one used in prognostication[3]: Type I: This is the classic, non-lethal type with autosomal dominant inheritance. The patients usually have blue sclerae, and the infants lack fractures at birth and are of normal height. 96% are able to walk, and 35 % are deaf, with the usual onset in childhood or puberty. Type II: This is perinatally lethal. Stillbirth or neonatal death is certain, making this a much more severe form of the disease with many fractures occurring from movement in utero and at birth. There is striking micromelia and bowing of extremities, the leg are held abducted at right angles to the body in the “frog-leg position.” Along with extremities defect, the other abnormality found in our case were facial abnormality, bluish sclera with bilateral proptosis, right sided inguinal hernia and congenital heart defect. Type II disorder is again subdivided into 3 type i.e. Type A is characterized by short, broad crumpled femora and continuously beaded ribs; Type B by short, crumpled femora but normal ribs or ribs with incomplete beading, and Type C by long, thin, inadequately molded, rectangular long bones with multiple fractures and thin, beaded ribs. Type A is due to a new autosomal dominant inheritance, so recurrence risk is 2-5%. A small thorax is seen with poor ossification of the skull and blue sclerae. Many of the fetuses will be small for gestational age. Types B & C contain recessively inherited forms. In our case, multiple fractures of the long bones along with thin beaded ribs was present, suggestive of type II OI with subtype C. Type III: This type is characterized by progressive deformity of the long bones and spine and often leads to an early death. The blue sclerae may fade or disappear later in life. Autosomal dominant and recessive inheritance is seen, with a recurrence rate quoted at 7%. Type IV: This is the mildest form and may not be separate from Type I. Inheritance is autosomal dominant and their sclerae can fade to white over time. Fractures and deformities are rare[4]. Skeletal manifestations are the hallmark of the OI group of disorders. Extraskeletal involvement may, however, contribute significantly to morbidity. Structural cardiovascular anomalies reported in OI include aortic root dilatation and aortic and mitral valve dysfunction[5]. Jánoskuti L reported a rare association of case of OI type I and membranous ventricular septal defect was presented in adult[6]. Manoria PC et al reported a case of OI with atrial septal defect[7]. In our case, the child diagnosed as type II OI along with atrial septal defect and right sided inguinal hernia. The diagnosis is possible by ultrasound examination between 16 and 24 weeks. Type II is usually apparent at less than 20 weeks. The appearance of normal long bones at less than 20 weeks does not eliminate the diagnosis, and serial measurements are recommended for diagnosis. Antenatally, lethality can be predicted at 14-16 weeks of gestation by ultrasonography with severe shortening of the long bones, femur length-abdominal circumference ratio of less than 0.16, hypoplastic thorax and marked bowing or fractures[1]. The diagnosis of OI congenita type II was based on the clinical findings of blue sclerae, craniotabes and multiple fractures at birth. A major finding in the congenital type of OI is multiple wormian bones in the skull particularly in the parietal, temporal and occipital regions. Congenital hypophosphatasia and battered baby syndromes are common differential diagnoses in the early neonatal period[8]. Decreased bone mass, disturbed organization and altered bone geometry resulting from abnormal collagen lead to bone fragility in OI. Molecular diagnosis depends on demonstration of abnormal incorporation of radioactive amino acids in collagen in fibroblast cultures. Genetic localization can be done by DNA probes[1]. Conclusion In conclusion, any case of OI should be screened for congenital cardiovascular defect and another abnormality. References
© Copyright 2008 - TUMS PUBLICATIONS The following images related to this document are available:Photo images[pe08029f1.jpg] |
| |||||||||

{kind=link}