 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
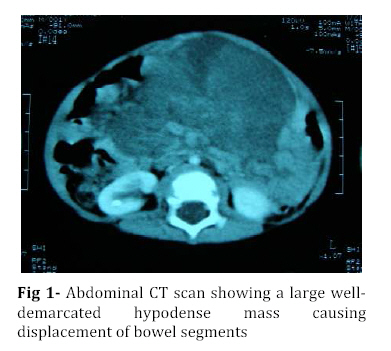
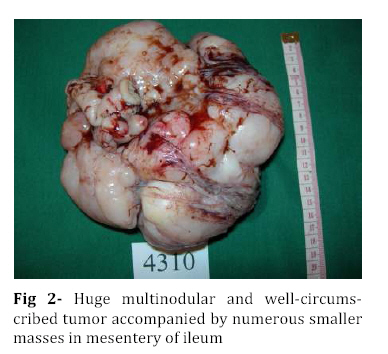
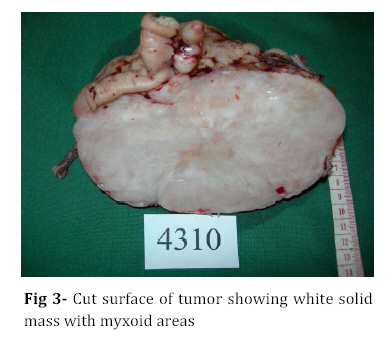
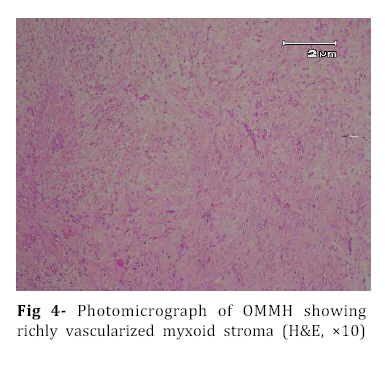
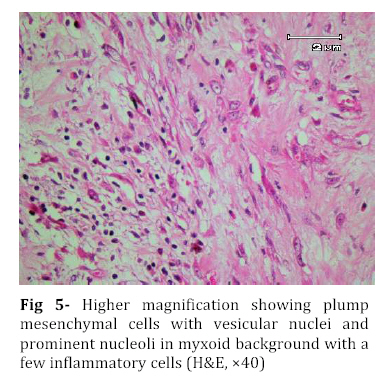
Iranian Journal of Pediatrics, Vol. 18, No. 3, Sept, 2008, pp. 273-276 Omental-Mesenteric Myxoid Hamartoma Mimicking Malignancy in a 14-month-old child (A Case Report) Nona Zabolinejad1, MD, Clinical Pathologist; Ahmad Bazrafshan2, MD, Pediatric Surgeon; Parya Dehghanian1, MD, Resident in Pathology; Naghmeh Zabolinejad, MD3, Resident in Dermatology 1. Department of Pathology, Mashhad University of Medical Sciences, IR Iran Code Number: pe08043 Background: The omental-mesenteric myxoid hamartoma (OMMH) is a very rare lesion, mainly seen in children and characterized by multiple omental and mesenteric nodules, which may be confused with malignant neoplasm. Microscopically, these lesions consist of a richly vascularized myxoid stroma with plump mesenchymal cells. This lesion has a benign clinical course without recurrence during follow up. Key Words: Omental-Mesenteric Myxoid Hamartoma; Inflammatory Myofibroblastic Tumor; Mesentery; Hamartoma; Abdominal mass; Childhood Introduction The omental-mesenteric myxoid hamartoma (OMMH) is a novel entity first described by Gonzalez-Crussi et al in 1983. They described three infants presented with multiple nodular tumors of the omentum and mesentery characterized histologically by plump mesenchymal cells in myxoid,well-vascularized stroma.Electron microscopy of one tumor revealed reticulated inclusions in dilated cisterna of endoplasmic reticulum.All of these patients survived with no evidence of disease following excision[1]. OMMH shares many morphologic features with inflammatory myofibroblastic tumor (IMT) and, in other words it may represent a variant of IMT[2]. These lesions may show deceptive features of immaturity and high cellularity that is apt to be confused with a true malignancy. If surgeons and pathologists are more familiar with this rare disease, the accurate diagnosis can be made earlier, thus avoiding some unnecessary postoperative options. Case Presentation A 14-month-old boy was referred to our hospital with history of abdominal distension, fever and vomiting for 3 months. Clinical examination revealed a tender mass at the midline of the lower abdomen. Ultrasonography showed a well defined hypoechoic mass lesion in the lower portion of abdomen and pelvic cavity which has caused moderate degree of hydronephrosis in the right kidney. Enhanced computed tomography (CT) revealed a huge well-demarcated hypodense and spherical mass which displaced bowel loops without obvious penetration to the intestinal walls (Fig 1). There was no paraaortic lymphadenopathy. The laboratory studies were all within normal limits. With suspicion of malignant neoplasm the patient underwent aspiration biopsy of the mass. Trucut biopsy showed a spindle cell lesion composed of small angular cells with small amount of eosinophilic cytoplasm. Some of cells had also plasmacytoid appearance. There was no evidence of necrosis, atypical changes or pleomorphism. Although a presumptive diagnosis of IMT was made but possibility of rhabdomyosarcoma couldn't be entirely ruled out. The patient underwent laparatomy; a huge multinodular, well-circumscribed tumor was found which was attached to the ileum with a clear boundary. The origin of the tumor seemed to be the mesentery. It was accompanied by numerous smaller nodules. The main tumor and the adjacent smaller nodules were completely removed with 7 cm margin of normal ileum (Fig 2). Gross examination revealed a huge (15×14 cm), multinodular white solid dominant mass with myxoid areas weighed 1190 gr which was localized in mesentery and accompanied by multiple smaller myxoid masses (Fig 3). We didn't find any invasion to the adjacent intestinal wall macroscopically. Histologically, tumor consisted of a richly vascularized myxoid stroma with plump mesenchymal cells containing vesicular nuclei and prominent nucleoli. A few inflammatory cells were also present (Fig 4, 5). There was no evidence of mitosis or necrosis. Immuno histochemmically, tumor cells were strongly positive for vimentin and desmin and were focally positive for smooth muscle actin. They were negative for EMA, NSE and MYO-D1. These findings confirmed the diagnosis of omental-mesenteric myxoid hamartoma. The patient had an uneventful postoperative course and was discharged from the hospital on the 10th post-operative day. No evidence of recurrence was noted during 3 years follow up.Discussion OMMH is a very rare lesion mainly seen during childhood[3]. To our knowledge, only ten cases, including ours, have been reported[4]. OMMH is considered a variant of IMT which is an umbrella term for nonspecific chronic inflammatory expansive lesions. There are variants of names for IMT including: plasma cell granuloma or pseudotumor, inflammatory myofibrohistiocytic proliferation, myofibroblastoma and OMMH[2]. There are three basic tissue patterns in IMT: a myxoid/vascular pattern, a compact spindle cell pattern, and a hypocellular fibrous pattern[4,5]. The clinical presentation is an abdominal mass accompanied by variable symptoms of malaise, anorexia, vomiting, and abdominal distension (the same as in our case)[6]. Recently a case of OMMH of the mesoappendix have been reported which had been incidentally detected after abdominal trauma[4]. The characteristic gross appearance of OMMH is a soft, lobulated pink or white circumscribed dominant mass with translucent and myxoid areas accompanied by numerous smaller myxoid masses throughout the peritoneum in a grape- like fashion[5,6]. Microscopically, these lesions consist of a richly vascularized myxoid stroma with plump basophilic mesenchymal cells having vesicular nuclei and prominent nucleoli. Cellular foci alternate with areas of collagenization. Occasionally, vacuolated and multinucleated cells are seen. The mitotic rate is low and the myxoid matrix contains acid mucopolysaccharides[6]. Mesenchymal cells are immunoreactive for vimentin,smooth muscle actin and sometimes for desmin and S-100 protein[4]. Ultrastructural examination reveals immature mesenchymal cells, fibroblasts, and vacuolated cells with dilated cisternae of rough endoplasmic reticulum and dense structures resembling lysosomes[1]. The presence of clonal chromosomal aberrations in this tumor; reported in a few studies, indicates that the OMMH is a neoplastic proliferation[2]. OMMH is not a clinically malignant neoplasm despite its cellularity, immature appearance, and resemblance to myxoid liposarcoma and other myxoid sarcomas, which has a practical importance in the clinical setting. Lipoblasts are not a feature of omental-mesenteric myxoid hamartoma and their absence distinguishes it from lipoblastomatosis[5]. Other tumors which may be considered in the differential diagnosis of (OMMH) are atypical gastrointestinal stromal tumor, inflammatory malignant fibrous histiocytoma, leiomyosarcoma, inflammatory cell-rich gastrointestinal autonomic nerve tumor, and solitary fibrous tumor[4]. However, the histological and immunohistochemical findings of our patient’s tumor did not suggest the above-mentioned diagnoses. Complete surgical resection remains the treatment of choice for OMMH. In all nine previous cases of OMMH, no recurrence was noted with favorable prognosis[4]. The same was true for our case. Although, all of the reported cases survived with no evidence of recurrence following excision[4], OMMH is considered a variant of IMT which may have an unpredictable course with local recurrences, and long-term postoperative follow-up is strongly recommended[7]. Conclusion Infantile lesions may show deceptive features of immaturity and high cellularity that are apt to be confused with a true malignancy. OMMH is a very rare lesion and because of its aggressive appearance, differential diagnosis with malignancy is warranted. The clinical picture of our case also led to high suspicion of malignancy. However by consideration of histological and immunohistochemical findings we could achieve the correct diagnosis and save the patient from unnecessary postoperative adjuvant treatments. References
© Copyright 2008 - TUMS PUBLICATIONS The following images related to this document are available:Photo images[pe08043f2.jpg] [pe08043f5.jpg] [pe08043f4.jpg] [pe08043f1.jpg] [pe08043f3.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}