 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
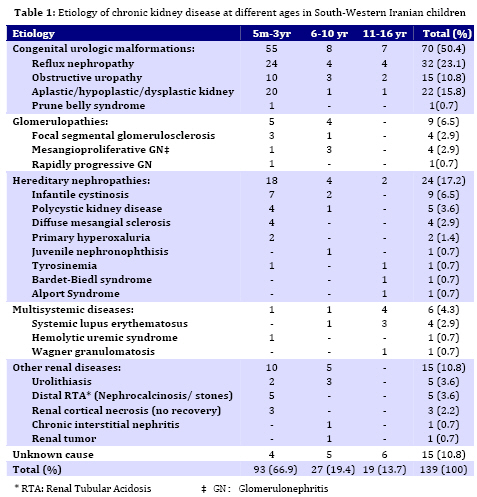
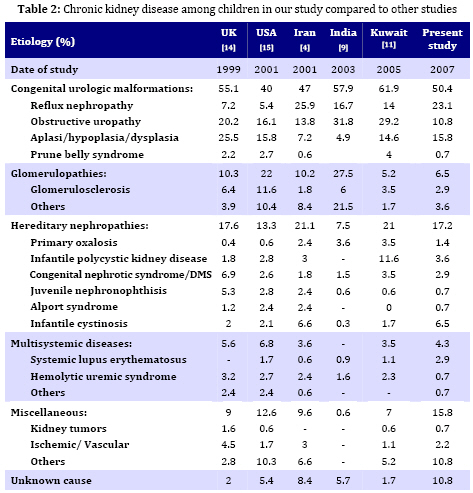
Iranian Journal of Pediatrics, Vol. 19, No. 2, June, 2009, pp. 147-153 Chronic Kidney Disease in Southwestern Iranian Children Ali Ahmadzadeh* 1, MD; Ehsan Valavi1, MD; Mehrnaz Zangeneh-Kamali1, MD; Azin Ahmadzadeh1, MD 1. Department of Pediatrics, Ahvaz University of Medical Sciences,Ahvaz, IR Iran Received: Sep 03, 2008; Final Revision: Dec 01, 2008; Accepted: Jan 23, 2009 Code Number: pe09023 Abstract Objective:The aim of the study was to determine the etiology of Chronic Kidney Disease (CKD) among children attending the pediatric nephrology service at Abuzar children's hospital in Ahvaz city, the referral center in Southwest of Iran. Key Words: Chronic renal failure; Obstructive uropathy; Reflux nephropathy Introduction CKD in children is the result of heterogeneous diseases of the kidney and urinary tract that range from common congenital malformations of the urinary tract, to rare inborn errors of metabolism that affect kidney function[1]. CKD is an irreversible condition that eventually progresses to end stage renal disease (ESRD). It is an important cause of morbidity and mortality in children worldwide [2,3]. The causes of CKD vary from one geographic area to another due to genetic and environmental factors. Some of these causes are preventable while in others, appropriate medical treatment and interventions may retard the progression of the disease [2]. In the absence of a national registry, there is paucity of information regarding the etiology of CKD in children from Iran[4]. An understanding of the causes of CKD is important as it may guide the distribution of limited resources towards its prevention. The aim of the present study was to determine retrospectively the etiology of CKD in children referred to our center. Subjects and Methods We reviewed the medical records of all patients diagnosed to have CKD at Abuzar children's medical center in Ahvaz, Iran, between April 1997 to May 2007. Clinical features including pallor, edema, oliguria, and hematuria were noted. Examination findings included weight, height and blood pressure. Pertinent laboratory data including blood chemistry, urinalysis, radiographic and scintigraphic studies and renal histopathology were recorded. CKD was defined as kidney damage or glomerular filtration rate (GFR) <60 ml/min/1.73 m2 as estimated by Schwartz`s formula[5] for 3 months or longer, regardless of the underlying etiology. The current definition encompasses all patients who were classified as having chronic renal insufficiency (CRI), chronic renal failure (CRF) and end stage renal disease (ESRD)[1]. The infants or children who had the mentioned criteria with at least a period of 3-month follow-up were included. The patients were excluded from the study if: (i) his or her follow-up period was less than 3 months; (ii) his or her information was incomplete. The etiological classification of CKD was as follows: Chronic glomerulonephritis (CGN) was defined clinically by irreversibility and histologically by obsolescence and sclerosis[6]. Hypertention was defined if the blood pressure (systolic or diastolic) levels were measured above 95th percentile + 5 mmHg for gender, age and height[7]. Growth retardation or failure to thrive (FTT) referred to growth less than the third or fifth percentile or change in growth that has crossed two major growth percentiles in a short time[8]. The underlying cause of CKD was considered to be reflux nephropathy in the presence of scarred kidney (irregular renal outline) demonstrated by ultrasonoraphy, intravenous pyelography or radionuclide and either of the following: (a) primary vesicoureteric reflux (VUR) demonstrated on voiding cystouretrography (VCUG) or radionuclide cystography, and (b) history and laboratory evidence of past urinary tract infections[9,10]. Obstructive uropathy was diagnosed if urinary tract dilatation was demonstrated by radiography or scintigraphy in the absence of VUR and bladder dysfunction. Neurogenic bladder was considered in patients with VCUG showing a large or a small bladder without any obstruction, bladder wall trabeculations and abnormal urodynamic studies (particularly in children over 5 years old). Diagnosis of renal hypoplasia and dysplasia was made on renal imaging (small kidney with regular outline with or without cysts) or characteristic renal biopsy. Polycystic kidney disease was diagnosed either on histopathology or ultrasonography (enlarged echogenic kidneys). Alport syndrome and juvenile nephronophthisis were diagnosed on characteristic renal biopsy with a positive family history. CRF was considered secondary to hemolytic uremic syndrome in patients with previous history of acute renal failure, microangiopathic hemolytic anemia and typical renal biopsy. Patients in whom the cause of CRF could not be identified were classified as unknown etiology. Findings A total of 139 children were included in the study over a 10-year period. There were 81 (58.2%) males and 58 (47.8%) females. The mean age at presentation of CKD was 4.2 (±3.6) years (range 3 months to 16 years). 93 (66.9%) children were below 5, 27 (19.4%) between 6 and 10, and 19 (13.7%) above 11 years old. The details of patients in different etiological groups and subgroups of each disease are summarized in Table 1. Obstructive uropathy was found in 15 (10.8%) patients. Boys were affected more commonly (70%) than girls. The mean age at presentation was 14 months. Failure to thrive (FTT) was present in 96% of the cases. Renal osteodystrophy was present in 28% and hypertension in 30%. Causes included posterior urethral valve in 4%, ureteropelvic junction obstruction or hydronephrosis in 1.4% of cases. Twenty-four (23%) patients had reflux nephropathy. The mean age at presentation was 4.6 years. Hypertension was present in 25.6%. Twenty-four patients (45 renal units) had asymmetrical renal scarring with a history of urinary tract infections in the past. CGN was diagnosed in 9 patients (6 girls, 3 boys). The mean age at presentation was 7.6 years. The diagnosis was established on renal biopsy. Focal segmental glomerulo-sclerosis and membranoproliferative glomerulonephritis were found each in 4 (2.9%) and crescent glomerulonephritis in one kidney. Lupus nephritis was seen in 4 patients. IgA nephropathy was not found. Renal dysplasia was found in 22 (15.8%) cases, presented at mean age of 5 months. These children were more stunted than the others. Infantile cystinosis was found in 9 (6.5%), polycystic kidney disease in 5 (3.6%) and diffuse mesangial sclerosis in 4 (2.9%) cases. These were the commonest causes of hereditary nephropathies. Alport syndrome, nephronophthisis and Bardet-Biedl syndrome were found each in one case. All the patients received standard treatment for CKD, including dietary modulation, calcium carbonate, active vitamin D analogues, iron and multivitamin supplements. Hypertension was treated with combination of calcium channel and beta-blockers, prazosin and loop diuretics. Anemia was treated by subcutaneous injections of erythropoietin. The mean duration of follow-up was 26 (±24.7) months. Peritoneal or hemodialysis was performed in 10 patients. Six patients underwent (4 live and 2 non-related) renal transplantation. The rest have died or received standard conservative management of CKD. Discussion It is important to know that the underlying causes for CKD are different in children than those seen in adults. Diabetic nephropathy and hypertension, dominant causes of CKD in adults, are very rare causes of CKD in childhood[1]. In Table 2, we compared our data with the data from the US, United Kingdom, Kuwait, India and a similar study in Iran. As a group, the leading causes of CKD in children are congenital and urologic anomalies, especially in the youngest age groups [1,3]. In the present study, CKD was more common in male infants; and 72% cases were younger than 12 months. CKD is a medical problem in pediatric population in south-west of Iran (Khuzestan province) and its neighboring Arab countries like Kuwait[11] and Saudi Arabia[12]. Genetic factors associated with consanguinity are important factors leading to a high incidence of hereditary diseases and congenital malformations. It is well-known that consanguinity is a common practice in Khuzestan province and most neighboring Arab countries. The precise incidence of CKD in Iran is not known. The age at presentation with the feature of CKD was lower than in India (8 years), and higher (4.2 years) as compared to reports from developed countries (74% higher than 5 years), suggesting delayed detection and referral of patients[10,13]. The etiology of CKD varies in different parts of the world. Hereditary disorders are common in regions where the frequency of consanguineous marriages is high[4,12]. Hereditary disorders including cystinosis, juvenile nephronophthisis, polycystic kidney disease, congenital nephrotic syndrome, primary hyperoxaluria, Bardet-Biedl syndrome were the second most common (17.2%) causes of CKD in our study and in a similar study performed in Tehran by Madani (21.1%)[4] . Preventable causes of CKD like obstructive uropathy and reflux nephropathy together accounted for majority of cases in our study, which is similar to a previous study from Iran and other parts of the world [4,10,16,17]. In our study, reflux nephropathy due to primary VUR was seen in 23.1% cases of CKD. However, the proportion of causes of CKD due to reflux nephropathy is much less in North American children, while no case has occurred in Swedish children from 1986-1994 [13,18]. In this study reflux nephropathy and obstructive uropathy were more common in males than in girls. Posterior urethral valve was the most common cause (86.6%) of urinary tract obstruction accounting for 13 (9.3%) of cases of CKD which was somewhat less than in Indian (14.7%) and other studies [10,17,18]. These conditions together contribute to 23-34% of CKD cases in developed countries. It is proposed that a decline in the proportion of patients with reflux nephropathy is chiefly due to prompt detection and management of urinary tract infections, followed by careful screening for underlying anomalies. Screening of urinary tract anomalies by antenatal ultrasonography is likely to detect significant structural disorders, which can be treated postnatal. Early and appropriate management of these disorders would prevent their progression to CKD [19]. Neurogenic bladder and secondary VUR were seen in 5.7% of patients, which is less than what Sirin et al reported [20]. In this study the proportion of patients with neural tube defect and secondary VUR was 15.4% in Turkish children. The proportion of patients presenting with ESRD (GFR <10 ml/min/1.72 m2) was higher (22%) in our study as compared to NAPRTCS report (4.3%) but lower than that reported by Gulati et al [17]. This again indicates late diagnosis and referral of patients to our center. The majority of our patients were anemic (Hb <10 g/dl), malnourished and stunted indicating an inadequate management of CKD. Stunting was more obvious in patients with obstructive uropathy and renal dysplasia than other conditions. Severe growth retardation in these patients could be attributed to early onset of CKD and tubular dysfunction (acidosis) in infancy[21]. Significant advances have been made in understanding various renal replacement measures, which have enabled provision of better care. Both chronic peritoneal dialysis and hemodialysis along with other supportive measures can ensure longevity and improved quality of life in patients of ESRD. However, chronic dialysis is, in the long- term, not able to achieve homeostasis and growth in children. So, kidney transplan-tation is considered the standard therapy for children with ESRD. Since prolonged dialysis is associated with multiple complications, it is usually advised, children with ESRD to undergo kidney transplantation as early as possible. Pre-emptive kidney transplantation, without prior dialysis is also encouraged in children. Conclusion A majority of cases of CKD in our region are due to obstructive uropathy and reflux nephropathy and may be preventable. Renal dysplasia is common in infants and toddlers, while CGN accounts for more cases of CRF in older children and adolescents. The majority of patients are referred late and only a few opt for renal replacement. Both these factors eventually lead to poor outcome of CKD in our population. Acknowledgment This study was supported by the vice-chancellery research of Jundishapour University of Medical Sciences. The authors would like to thank Mr. Charaghian for his help in statistical analysis of the results. References
© 2009 by Center of Excellence for Pediatrics, Children’s Medical Center, Tehran University of Medical Sciences, All rights reserved. The following images related to this document are available:Photo images[pe09023t2.jpg] [pe09023t1.jpg] |
| |||||||||

{kind=link}
{kind=link}