 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
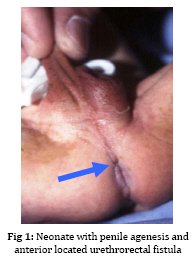
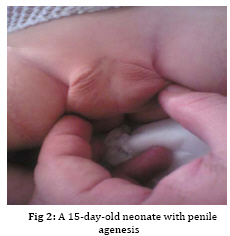
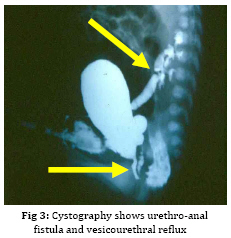
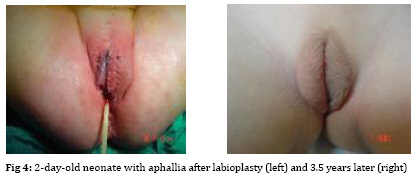
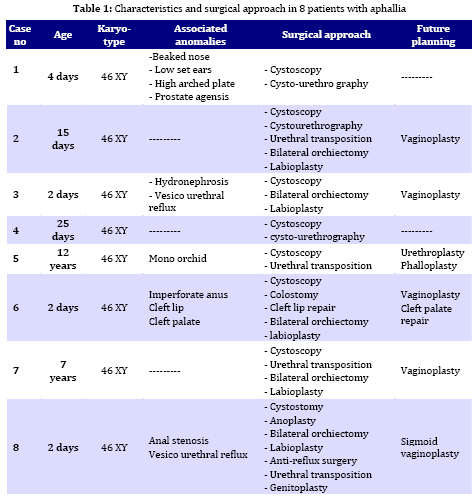
Iranian Journal of Pediatrics, Vol. 19, No. 2, June, 2009, pp. 173-179 Penile agenesis: Report on 8 Cases and Review of Literature Alireza mirshemirani*1, MD; Ahmad Khaleghnejad1, MD; Hoshang Pourang2, MD; Naser Sadeghian1, MD; Mohsen Rouzrokh1, MD; Shadab Salehpour3, MD 1. Pediatric Surgery Research Center, Shahid Beheshti University of Medical Sciences, Tehran, IR iran Received: Jun 27, 2008; Final Revision: Dec 01, 2008; Accepted: Jan 14, 2009 Code Number: pe09028 Abstract Background: Penile agenesis (PA) is an extremely rare anomaly with profound urological and psychological consequences. The opening of the urethra could be either over the pubis or at any point on perineum or most frequently in anterior wall of the rectum. The aim of treatment is an early female gender assignment and feminizing reconstruction of the perineum. Key Words: Aphallia; penile agenesis; reconstruction; Ambiguous genitalia Introduction Agenesis of the penis which has a reported incidence occurring once in 30 million live births, is an extremely rare genitourinary anomaly [1,2]. Penile agenesis (pa) is believed to result from either the absence of the genital tubercle or its failure to develop[3]. Several investigations claim the absence of corpora cavernosa and corpus spongiosum as a prerequisite for the diagnosis of PA[4]. Patients usually have 46XY karyotype [5]. There are two major groups of patients with pa, those with congenital anomalies incompatible with life and those with pa as solitary malformation[6,7]. More than half of these patients have associated anomalies including develop-mental defects of the caudal axis, genitourinary (54%) and gastrointestinal tract anomalies[8]. Pa as solitary malformation consists of absent penis and usually normal appearingg scrotum which contains palpable testicles with normal function[3,7,9,10]. Skoog and bellman[8] classified patients based on the relationship of the urethral meatus to the anal sphincter. The three variations they described are postsphinctric with anterior perianal urethra, presphincteric with urethrorectal fistula, and urethral atresia with vesicorectal fistula. The opening of the urethra could be either over the pubis or at any point on the perineum or, most frequently, in anterior wall of the rectum[8,11]. the original description originates from imminger in 1853. Since then approximately 80 cases have been reported worldwide[6,7]. Treatment for these patients is controversial. Surgical intervention during infancy, including urethral transposition and bowel vaginoplasty via a posterior sagittal approach plus orchiectomy and scrotoplasty, has been advocated. However, gender reassignment in other contexts and detailed psychosexual data may not support this recommendation[8]. In addition, since phallic reconstruction is feasible, if imperfect, in prepubertal boys maintenance of male gender should be considered an option in discussions with the patient’s family[9]. Case(s) Presentation Case 1:a 4-day-old neonate, 2.1 KG birth weight, with aphallia and urination and defecation through rectum. Physical examination revealed absence of penis, apparently normal scrotum, and bilateral well descended testes. Anus was located on its real place and urethraopened through the anus (Fig. 1). The nose wasbeaked and he had Low-set ears and high-arched palate. Karyotype 46XY and HCG trial test showed normal testosterone production ability of the testes. cystourethrography was performed under low general anesthesia which showed an anterior located urethrorectal fistula. There was a congenital agenesis of prostate as associated anomaly. Unfortunately parents refused any surgical intervention. Case 2: A 15-day-old neonate was referred to us due to absence of penis and urination and defecation through rectum. He had normal scrotum, bilateral normal testis position, normally located anus and urethral opening through anus (Fig. 2). Ultrasonography showed no associated anomalies. Buccal smear was consistent with male genotype, and chromosomal studies revealed a normal karyotype 46Xy; laboratory tests were unremarkable. Cystourethrogram performed through rectosigmoidoscopy showed normal appearing bladder, and an urethrorectal fistula opening proximally into the rectum. Treatment: Surgical intervention included urethral transposition, bilateral orchiectomy and labioplasty. A later labio-vaginoplasty was planned. Case 3: a 2 days old neonate, with 2900 kg birth weight, abdominal distesion, vomiting and no urination and defication. Patient had aphallia, normal scrotum and bilateral normal testis postion, and urethra at anterior anus position, buccal smear was consistent with male genotype, and chromosomal studies revealed a normal karyotype of 46XY. Ultrasonography and renal scan revealed hydronephrotic kidney with nearly normal function, and cystogram showed urethro-anal fistula and vesicourethral reflux (Fig. 3). Treatment: Urgent cystostomy due to elevated bun at 4th day of life, bilateral orchiectomy and labioplasty at 20 days. Because of uti patient was discharged under coverage of antibiotherapy to be followed up. Case 4: A 25-day-old neonate with aphallia, normal scrotum and bilateral testis, urination from anterior site of the anus, karyotyp 46XY, without any associated anomalies. His parents refused any surgical intervention. Case 5: 12-year-old patient with aphallia, normal scrotum, testis normal in the left and absent in the right side; 46XY karyotype and urethra opening in anterior anus site. Treatment: Cystoscopy via urethral fistula, urethral transposition to the lower scrotal area. As patient and his parents would prefer male gender, so he was discharged and advised to come later for masculinizing operations (urethroplasty and penile reconstruction). Case 6: 2-day-old neonate was admitted for imperforate anus, aphallia, normal scrotum, normal bilateral testes, 46XY karyotype, urethral opening at the site of mid-scrotal area, and cleft lip and palate. Treatment: Sigmoid colostomy, bilateral orchiectomy, labioplasty and repair of cleft lip and planning for later vaginoplasty and repair of cleft palate. Case 7: 7-year-old patient with aphallia, normal scrotum, normal bilateral testes, 46XY karyotype, urethra opening at the anterior anal site, and no associated anolmalies. Treatment: Urethral transposition to the scrotal area, bilateral orchiectomy, labioplasty and planning for later vaginoplasty. Case 8: 2-day-old neonate with aphallia, anal stenosis, normal scrotum, normal bilateral testis, 46XY karyotype, urine passage from anus. Urethra cystography showed grade II vesico-urethral reflux. Trertment: anoplasty and cystostomy at 12th day of life, bilateral orchiectomy at 5 months, anti-reflux surgery and transposition of urethra toperineal area at one year, genitoplasty at the age of 3 years, repositioning of urethra to more anterior perineal site at 5 years. Patient is now 6.5 years old and is candidate for sigmoid-vaginoplasty at a later age (Fig. 4). Discussion Congenital absence of the penis is a rare form of ambiguous genitalia [12,13]. This anomaly was first described by imminger in 1853. Since then approximately 80 cases have been reported worldwide [6,7]. Development of the caudal axis is initiated early in embryonic life. During week 3 of embryogenesis, mesodermal tissue from the region of the primitive streak migrates down about the cloacal membrane to form the cloacal folds. These folds unite in front of the cloacal membrane and form genital tubercle. While this occurs externally, the cloaca is divided internally into the anterior urogenital sinus and posterior rectum by the urorectal septum. As the embryo matures, the genital tubercle elongates and the cloacal folds divide into the anterior urethral and posterior anal folds. The genital swelling appears at this time on either side of the cloacal folds, these swellings eventually will migrate caudally, coming together over the urethral folds and fusing at the median raphe to form the scrotum. The genital tubercle will fuse to form the urethra internally and the penis with its median raphe externally [2,14,15]. penile agenesis is believed to be due to deficient formation of the genital tubercle or its failure to develop into a penis in the fourth week of embryogenesis [12,13].this hypothesis seems to hold true in the usual case of pa with normal scrotal formation, normal median raphe development and perforate anus. In such situations all structures of the caudal axis are preserved except for the penis itself[14].in most cases the urethral opening is located either on the perineum between the scrotum and anus or as a fistula to the gastrointestinal tract, typically to the rectum [8,10,16]. Skoog and belman[8] suggested three variants, based on urethral position in relationship to the anal sphincter, as postsphincteric, presphincteric (prostato-rectal fistula) and urethral atresia. More proximal the bladder outlet, greater is the likelihood of other anomalies and death [8]. The diagnosis of this abnormality, includes: complete absence of corpora cavernosa and corpus spongiosum and opening of the urethra in perineum near the anus or into the rectum [17,18]. pa must be differentiated from concealed penis, rudimentary penis, micropenis, male pseudohermaphroditism, and intrauterine amputation of the penis[12]. As for treatment, if the case was brought in infancy, feminizing operations are indicated[18,19]. For the cases brought after the second year of life, as their sexual identification is appeared, it is advised to perform masculinizing operations, in order not to disturb the patient psychologically[4,7-9], as we have done it in one of our cases. Penile reconstruction among children has been a controversial subject now and is a challenge in pediatric surgery [9,20]. Gilbert et al [9] have performed penile reconstruction on seven children, perovic[20] performed on 5 children, akoz et al [6] performed on two children. Children's penis reconstruction depends on age in time of operation and length of neophallus and is still a matter of controversy [6,9]. The present consensus is that patients affected by aphallia are better raised as males[21]. Recently, successful phallic reconstruction in two patients with aphallia were presented from bologna (italy) using the lower abdominal wall skin flap for making the shaft and the bladder/labial mucosa free graft for making the urethra[22]. The procedures were completed at 9 and 17 months of age. However, all is not that well. Male genitoplasty in such cases requires a long urethroplasty and is not supported by corpus spongiosum. Due to lack of urethral resistance, the long and the short term results of the long urethroplasty are not satisfactory. Patients are not able to expel urine at the meatus in a forceful stream. This problem was encountered in one of the two cases from bologna and a scrotal urethrostomy had to be resorted to later on. We also had the similar experience in a case in whom male genitoplasty was performed [23]. the initial management should include dismantling of the urethra from the anorectum and placing the same in the perineum (as preliminary urethro-stomy)[23]. This may be done even during the newborn period or infancy so as to avoid urinary complications in future. The child should not be discharged too early without a urethrostomy as death due to chronic renal failure has been reported[23].If the parents arenot sure of the gender, they may be given time to think it over [23]. We have reviewed 8 cases of penile agenesis from 3 pediatric surgical units in Tehran. 2 cases were hospitalized and after evaluation refused our advice and left the hospital, but in 6 cases we performed different surgical interventions. In our cases only one patient had mid-scrotal type of urethral opening. 5 patients were changed to female gender as mentioned before, and one patient tended to have male gender. All the six patients were planned for final surgical approach. Some associated anomalies were repaired too. Conclusion In newborn period or infancy, feminizing operations are recommended for treatment of PA, but after two years, as sexual identification of the patients has appeared, it is advised to perform masculinizing operations in order not to disturb the patients psychologically, and finally, no surgical intervention should be performed before counseling the parents. Acknowledgment The authors would like to thank Mrs. M Saeedi for her kind assistance in preparing the manuscript. References
© 2009 by Center of Excellence for Pediatrics, Children’s Medical Center, Tehran University of Medical Sciences, All rights reserved. The following images related to this document are available:Photo images[pe09028t1.jpg] [pe09028f3.jpg] [pe09028f4.jpg] [pe09028f2.jpg] [pe09028f1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}