 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
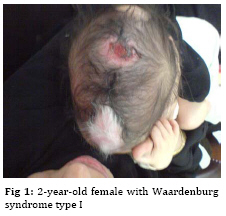
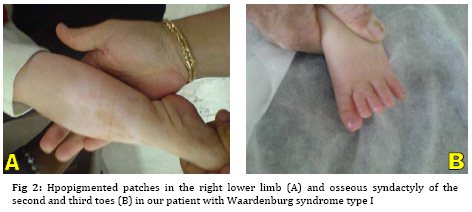
Iranian Journal of Pediatrics, Vol. 19, No. 2, June, 2009, pp. 189-192 Waardenburg Syndrome Type I in an Iranian Female Naeimeh Tayebi 1, MD 1. Genetic Counselor, Genetic Research Center, Shahid Fiazbakhsh Rehabilitation Comprehensive Center, Welfare Organization, Yazd, Iran Received: Jul 11, 2008; Final Revision: Nov 10, 2008; Accepted: Dec 12, 2008 Code Number: pe09031 Abstract Background:Waardenburg syndrome (WS) is a rare, autosomal dominant disorder characterized by congenital hearing loss; dystopia canthorum; broad nasal root; depigmantation of hair, skin or both; and heterochromic iris. WS is classified into four types, WS1, WS2, WS3 and WS4. In this paper, we report a new case of Waardenburg syndrome type I in an Iranian female. Key Words: Waardenburg syndrome; White forelock; Congenital sensorineural hearing loss Introduction Waardenburg syndrome (WS) is a rare, autosomal dominant disorder characterized by congenital hearing loss; dystopia canthorum; broad nasal root; depigmantation of hair (white forelock), skin or both; and heterochromic or hypochromic iris. This condition is caused by the physical absence of melanocytes in the skin, hair and eyes. Also, this syndrome is classified as a disorder of neural crest cell development [1]. The prevalence of this syndrome is estimated to be between 1 in 10.000 and 1 in 50.000. In addition, Waardenburg syndrome equally affects both sexes and all races[2]. Depending on additional symptoms, WS is classified into four types, WS1, WS2, WS3 and WS4. Type 1 is the classic form of WS with dystopia canthorum (lateral displacement of the inner canthi), type 2 characterized by the presence of white forelock, unilateral or bilateral deafness, but without the dystopia canthorum, type 3 is pseudo WS and type 4 is with Hirschsprung disease [3]. The manifestations of WS can vary widely among individuals, even those of the same family. One of the most striking features of WS is a white forelock which is found in about 45% of affected individuals. Hypopigmentation of the skin is less frequent than that of the hair, but white patches are sometimes observed on the trunk, face and limbs [4]. Congenital sensorineural hearing loss is found in about 60% of individuals with Waardenburg syndrome. This deafness is a present feature in approximately 25 % of WS I and in 50 % of WS II. The loss can be moderate to profound. Also, it may be unilateral or bilateral and the laterality can vary among family members [5]. Occasionally, spina bifida, cleft lip and palate have been observed in this syndrome [4]. The diagnosis of WS is made clinically. Genetic testing is available for confirmation of diagnosis and prenatal diagnosis if the mutation in the family has been identified [6]. In over 90% of individuals with WS a mutation was identified in the PAX3 gene. This gene, located on chromosome 2 is involved in the regulation of melanocyte development [7]. In this paper, we report a new case of Waardenburg syndrome type I in Iran which has been investigated at the Genetic department of Yazd Welfare Organization. Case presentation A 2-year-old full term female was referred to genetic research center of Welfare Organization, Yazd, Iran, in July 2008 because of congenital deafness and a white forelock. She was the first child of non consanguineous parents. Her prenatal, natal and post natal history was unremarkable. Her height, weight and head circumference at birth and admission were normal. Her family history revealed that her father was similarly affected. Her developmental milestones were normal. Also, as is shown in Figure 1, the skin of skull was not formed. With growing hair, central white forelock gradually appeared. The nasal root was hypertrophied. There was dystopia canthorum (lateral displacement of medial canthi) and telecanthus. Eye examination was normal and no heterochromia was seen. Also, accommodative isotropic in both eyes were seen. Profound unilateral sensory neural hearing loss (left ear) was noted on auditory brainstem response examination. Hpopigmented patches were observed in the right lower limb (Figure 2A). Osseous syndactyly of the second and third toes was seen (Figure 2B). The joints showed no deformity. The external genitalia were unremarkable. On Paraclinical examination, G-banded chromosome study demonstrated a normal karyotype, 46 XX. Also, routine biochemistry studies, abdominal ultrasound and brain MRI were described as normal. Echocardiogram showed small perimembrane VSD. Discussion Waardenburg syndrome is a hereditary auditory pigmentary syndrome. It was described in 1950 by Waardenburg [8]. There are five major and five minor diagnostic criteria for WS. Major criteria include sensorineural hearing loss, iris pigmentary abnormality, hair hypopigmentation (white forelock or white hairs at other sites of the body), dystopia canthorum and first-degree relative previously diagnosed with Waardenburg syndrome. Minor criteria include skin hypopigmentation (white skin patches), broad nasal root, hypoplastic alae nasi, medial eyebrow flare and premature graying of the hair before age 30. An individual must have two major or one major plus two minor criteria to be considered for diagnosis of WS[9]. Our case was the first child of non consanguineous parents, her mother was healthy but her father had signs like her, hence the inheritance of this syndrome in this case is autosomal dominant. Also, she had unilateral profound sensorineural hearing loss, white forelock, dystopia canthorum, broad nasal root, hypopig-mentation of the skin and scalp defect without iris heterochromia or hypochromia. The scalp defect is a new sign in this syndrome and it has not been reported until now. Moreover, this Patient did not show signs of abdominal distension; as a result, she is a new case of WS type I. WS has been classified into four distinct subtypes [10]. Dystopia canthorum is the most distinguishing feature of WS type 1[3]. In 2005, Otman et al reported WS type 2 in an African patient with congenital deafness, white forelock, variable sized hypopig-mented patches over the whole body, wide nasal root, iris heterochromia, but no dystopia canthorum[11]. Egbalian described a case of Waardenburg-Shah syndrome (WS4) in a five-day-old female newborn with signs of abdominal distension, white forelock with poliosis of both the upper and lower eyelashes of eyes and medial eyebrow, broad nasal root and blue iris[12]. Folic acid supplementation in pregnancy has been recommended for women at increased risk of having a child with Waardenburg syndrome[13]. Conclusion We report a case of Waardenburg syndrome type I with features such as unilateral profound sensorineural hearing loss, white forelock, dystopia canthorum, broad nasal root, hypopigmentation of the skin and scalp defect without iris heterochromia or hypochromia. Acknowledgment This research was granted by Genetic Research Center, Shaid Faiazbakhsh Rehabilitation Comprehensive Center, Yazd Welfare Organization, Iran. The author expresses her thanks to Mr. Hossein Khodaee for helping in cytogenetic analysis. References
The following images related to this document are available:Photo images[pe09031f1.jpg] [pe09031f2.jpg] |
| |||||||||

{kind=link}
{kind=link}