 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
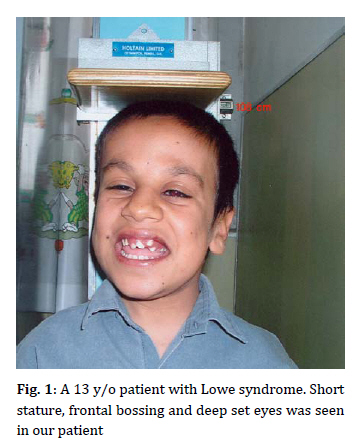
Iranian Journal of Pediatrics, Vol. 19, No. 4, 2009, pp. 417-420 ♀Lowe Syndrome: Report of a Case and Brief Literature Review Gholamhossein Amirhakimi*1, MD; Mohamad-Hosein Fallahzadeh1, MD; Hedyeh Saneifard1, MD Department of Pediatrics, Shiraz University of Medical Sciences, Shiraz, IR Iran * Corresponding Author; Address: Nemazee Hospital, Shiraz, IR Iran E-mail: amirhakg@sums.ac.ir Received: Jul 20, 2008; Final Revision: Jun 03, 2009; Accepted: May 01, 2009 Code Number: pe09052 Abstract Background: The oculocerebrorenal syndrome of Lowe (OCRL) is a rare x-linked recessive disorder first described in 1952. This syndrome is characterized by ocular involvement, mental retardation and kidney disease. The causative gene is OCRL1. Survival rarely exceeds 40 years. Key Words: Cataract; Hypotonia; Renal tubular acidosis; Mental retardation; Short stature; Oculocerebrorenal syndrome Introduction The oculocerebrorenal syndrome of Lowe (OCRL) also referred as the Lowe syndrome is a rare disorder distinguished by a triad of organ system abnormalities, namely ocular disease such as neonatal onset cataracts, mental retardation and renal dysfunction [1]. It was described in 1952 by Lowe and colleagues[2]. The prevalence of this syndrome has been estimated to occur in 1 out of 500000 individuals [3]. OCRL results from a mutation in the oculocerebrorenal gene (OCRL1) being localized on Xq24-26.1 that encode a protein highly homologous to inositol polyphosphate 5-phosphatase. This suggests that Lowe syndrome may represent an inborn error of inositol phosphate metabolism [4]. Presence of eye, central nervous system and kidney involvements is required for the diagnosis of this syndrome [6]. Congenital bilateral cataract is present at birth in all patients. Other ocular defects include glaucoma and searching nystagmus[7]. Hypotonia and neonatal areflexia are present. Motor and mental development are delayed and stereotypic behaviors, such as temper tantrums and aggressiveness is frequent during adolescence [3]. Seizures may accompany the disease and dermal findings of a cystic nature is reported in a few cases with this syndrome [2]. Renal Fanconi syndrome may present in the first months of life and differ in severity between individuals [8]. Facial dysmorphism is often present and consists of frontal bossing, deep set eyes, chubby cheeks and fair complexion[9]. Dental findings include prolonged retention of primary teeth, chronic subrachitic state, enlarged pulp chambers and mildly dysplastic dentin formation [10,11]. Case Presentation A 13-year-old boy was referred with short stature (Fig 1). There was no history of parental consanguinity or affected siblings in the family. There were 3 sisters with normal stature and intelligence. He had psychomotor retardation; smiling at one year of age and sitting at 2 years. He had history of congenital bilateral cataract for which lensectomy was done. Around the 8th month of age he had facial puffiness for which he was admitted to the hospital and the diagnosis of renal tubular acidosis was made. There was history of unilateral undescended testis. In physical examination his weight was 21 kg, height 108.2 cm, and head circumference 52.5 cm. He had frontal bossing, deep set eyes, chubby cheeks, maxillary prognatism, chronic gingivitis and malocclusion. In neuroocular exam-ination, he had searching nystagmus. In neurologic examination he had areflexia. Mental retardation was noted and he had aggressive behaviors, irritability and outburst of anger. He had renal tubular acidosis, (Fanconi type) for which he was followed by nephrologist. Discussion The oculocerebrorenal syndrome of Lowe is a rare multisystem disorder primarily affecting the eyes, brain and kidney. Only 190 patients have been identified in the year 2000[3]. Various disturbances of tooth eruption have been found with the OCRL syndrome notably over-retention of primary teeth and delayed eruption of permanent teeth [1,12]. Dense cataract is present at birth in all patients. Glaucoma (present in 50% of patients, with or without buphthalmus) is detected within the first year of life or even later. Sight sharpness is compromised, aphakia together with retinal dysfunction is responsible for nystagmus[6,7]. In central nervous system examination, severe hypotonia often with absence of DTRs are present at birth. Motor development is retarded and the autonomous gait becomes apparent generally after the third year. Moderate to severe mental retardation with IQ of 50 or less is present in 10% of patients. 87% of patients show evidence of conduct disturbance with auto and heteroaggressiveness, irritability, outbursts of anger and non finalized behavior. 80% of patients over 18 years old have seizures [2,3]. Renal disease is primarily characterized by renal Fanconi syndrome. The severity of the renal disease can vary significantly between patients and tends to worsen with age. At birth many children are asymptomatic. The first symptoms generally develop during the first months of life and are generally related to renal bicarbonate, salt and water wasting, causing failure to thrive[8,13]. In the present patient congenital bilateral cataract was present at birth for which lensectomy was done, searching nystagmus was also observed in ocular examination. Hypotonia was present and he had motor and mental retardation, irritability and aggressiveness. At 8 months of age renal Fanconi syndrome was diagnosed. He had history of unilateral undescended testis. Brook et al reported a boy with Lowe syndrome and multiple unerrupted teeth and pericoronal radiolucencies[1]. Harrison and coworkers reported a child with this syndrome who had generalized mobility of all primary teeth at the age of 4 years[10]. Ruellas and coworkers also reported an 18-year-old boy with Lowe syndrome and dental anomalies [11]. Another case was a 3-year-old boy with Lowe syndrome who previously underwent bilateral cataract surgery and strabismus surgery in Neworlean[7]. Wang and coworkers also reported the first case of Lowe syndrome in Taiwan, a newborn who was born with congenital cataract, glaucoma, generalized hypotonia with areflexia and then developed renal tubular dysfunction[6]. Another observation was in a boy with history of congenital cataracts and mild developmental delay was also found hematuria with proteinuria but minimal signs of renal tubular dysfunction reported by Andera et al[13]. At birth ocular involvement with bilateral cataract and hypotonia may be found in congenital infections (rubella), peroxisomal disorders, mitochondropathies, myotonic dystrophies or congenital myopathies (Muscle Eye Brain disease). The appearance of renal involvement excludes these alternative diagnoses within the first months of life[3]. The disease is caused by reduction of phosphatidylinositol biphosphatase activity below 10% in fibroblasts[6,9]. Female carriers of Lowe syndrome may be detected in 94% of the cases by slit lamp examination because of significant punctate white to gray opacities and disturbance in a radial fashion in all layers of the lenticular cortex. Antenatal diagnosis is made by enzymatic activity in cultured chorionic villi at 9-11 weeks or in cultured amniotic fluid cells (15-20 weeks) [4]. Management of the disease includes early removal of cataract to avoid amblyopia. Ocular tone has to be tested frequently in order to diagnose glaucoma. Early targeted rehabilitation therapy is necessary to treat hypotonia. Areflexia does not require treatment. Seizures require treatment with drugs specific for symptoms. Drugs such as neuroleptics, stimulants, benzodiazepins, anti depressives (tricycle antidepressants and serotonin reuptake inhibitors) although adequately prescribed, are only partially efficacious [2,3]. Renal tubular acidosis must be recognized and treated promptly with alkali supplements to maintain serum bicarbonate level at around 20 mEq/L. Potassium citrate is particularly useful as it also helps to prevent nephrocalcinosis and tends to reduce renal calcium excretion. Rickets should be treated with oral phosphate supplements and vitamin D. Treatment should be targeted towards maintaining serum calcium and PTH level within normal range and serum phosphate levels above 3 mg/dl. Cryptorchidism may improve spontaneously and surgery is rarely required [3]. Conclusion The possibility of OCRL should be considered in boys with cataracts and glumerolar disease. Historically the life span of affected patients extended only to the second or third decade with the causes of death related to the respiratory illness, epileptic seizures and renal failure. Recently increased adult survival has been attributed to the implementation of aggressive medical care. The quality of life depends on the extension of the mental and renal manifestations and specific treatment which is provided for the patients. References
© Copyright 2009 - TUMS PUBLICATIONS The following images related to this document are available:Photo images[pe09052f1.jpg] |
| |||||||||

{kind=link}