 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Iranian Journal of Pediatrics, Vol. 20, No. 3, July-September, 2010, pp. 358-362 Ehlers-Danlos Syndrome Type VI in a 17-Year-Old Iranian Boy with Severe Muscular Weakness; A Diagnostic Challenge? Ariana Kariminejad1, MD; Bita Bozorgmehr1, MD; Ali-Reza Khatami2; MD; Mohamad-Hasan Kariminejad1, MD; Cecilia Giunta3, MD, and Beat Steinmann3, MD 1Kariminejad
Najmabadi Pathology and Genetics Center, Tehran, IR Iran Received: May 20, 2009; Final Revision: Nov 14, 2009; Accepted: Jan 18, 2010 Code Number: pe10054 Abstract Background: The
Ehlers-Danlos syndrome type VI (EDSVI) is an autosomal recessive connective
tissue disease which is characterized by severe hypotonia at birth, progressive
kyphoscoliosis, skin hyperelasticity and fragility, joint hypermobility
and (sub-)luxations, microcornea, rupture of arteries and the eye globe,
and osteopenia. The enzyme collagen lysyl hydroxylase (LH1) is deficient
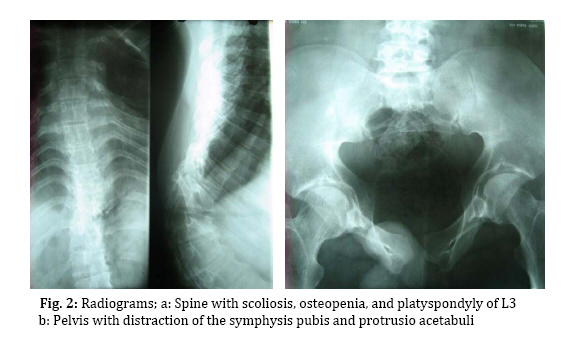
in these patients due to mutations in the PLOD1 gene. Key Words: Ehlers-Danlos; Kyphoscoliosis; Muscular hypotonia; Muscular weakness; Microcornea Introduction The Ehlers-Danlos syndrome is a heterogeneous group of inherited connective tissue disorders characterized by skin hyperelasticity and fragility, and joint hypermobility. They have been classified into types I (OMIM 130000), II (OMIM 130010), III (OMIM 130020), IV (OMIM 130050), VIIA and VIIB (OMIM 130060) with autosomal dominant inheritance and types VI (OMIM 225400), VIIC (OMIM 225410), and other types (OMIM 606408 and 612350) with autosomal recessive inheritance[1,2,3]. The Ehlers-Danlos syndrome type VI (EDS VI), also called the kyphoscoliotic type[1], is characterized by severe hypotonia at birth, joint laxity, hyperextensible and fragile skin, severe progressive kyphoscoliosis, osteopenia, and rupture of arteries and of the eye globe[1,2]. The exact prevalence of EDS VI is unknown but estimated to be world-wide approximately 1:100,000 live births[4]. EDS VI is caused by deficient lysyl hydroxylase (LH1)[5], an important posttranslational modifying enzyme of collagen synthesis due to mutations in the PLOD1 gene[6]. This enzyme catalyzes the hydroxylation of collagen lysyl residues to hydroxylysyl residues which are essential for the formation of stable collagen cross links, thus confining collagen its tensile strength; furthermore they are necessary for the attachment of galactosyl and glucosyl-galactosyl residues. A second, rarer form of EDS VI has been described, EDS VIB (as opposed to EDS VIA), in which patients have a similar clinical phenotype of EDS VI but with normal levels of LH1 activity, absence of mutations in PLOD1, and normal cross links. Deficiency of LH1 gives rise to an abnormal pattern of lysyl pyridinoline (LP) and hydroxylysyl pyridinoline (HP) cross links formed in vivo and excreted in the urine[7]. The ratio of urinary total LP to HP in patients with EDS VI is much higher compared to normal controls[2,3,8]. Hence, we can take advantage of this finding and use it as an inexpensive tool for the diagnosis of EDS VI. At least 30 different mutations in the PLOD1 gene have been identified (Giunta et al, in preparation). Here we report a 17-year-old boy with typical clinical features, and biochemical and molecular findings of EDS VI, and emphasize the differential diagnosis of muscular disorders. Case Presentation This 17-year old boy is the first child of cousin parents once removed. The second and third children, a boy and girl, died at 1 year and 5 days of age, respectively, of unrelated cause. He was admitted to us for the evaluation of his muscular weakness. Pregnancy, delivery at term, and neonatal adaptation were uneventful; his weight was 3,000 grams but length and head circumference were not recorded. He developed hyperbilirubinemia during the first week after birth which required blood transfusion. Severe hypotonia was noticed since birth and kyphoscoliosis developed during the first months of life. Because of muscular weakness he had to be bottle-fed with a large-holed teat. Gross motor development milestones were delayed. He could stand without support at 2 years and started to walk at 2.5 years. He also had speech delay and could say his first words at the age of 20 months. He went to school for 5 years but did not continue because of his motor disabilities. Although intelligence has not been formally evaluated, based on his relative good performance at school we believe that there is no mental retardation. He had a history of rupture of the ocular globe at age 15 years after his father's elbow accidentally hit his face, and of multiple dislocations of both wrists and ankles. On physical examination at the age of 17, his height, weight and head circumference were 165 cm, 70 kg, and 55 cm, respectively. He had down-slanting palpebral fissures (Fig. 1a), microcornea (9 mm in diameter as opposed to 11-12.5 mm in controls[2]), ptosis, prosthetic left eye, and slightly bluish sclera. The skin was soft, with multiple scars on knees due to poor healing, and with hyperkeratosis follicularis (Fig. 1c). He also had hypermobile joints; however, hyperelasticity of the skin was not prominent. Generalized hypotonia was noted and muscle strength was decreased. Muscle force was estimated to be 3-4 (had active movement against gravity and some movement against resistance); however, reflexes and muscle bulk were normal. Skeletal survey revealed osteopenia, kyphoscoliosis, and platyspondyly of L3 (Fig. 2a) as well as distraction of the symphysis pubis and protrusio acetabuli (Fig. 2b), and downward slanting and thinning of the ribs (not shown). Bone mineral density (Norland XR-36) was normal in spine (0.991 mg/cm2) and hip (0.991 mg/cm2) but decreased in the femur (i.e., 0.810 mg/cm2 which is 1.2 SD below the age-matched reference). Creatine phosphokinase, electromyogram, and nerve conduction velocity were normal. Urine samples from the patient and parents were analyzed for urinary pyridinolines as reported[8]. The ratio of urinary total pyridinolines, lysyl pyridinoline to hydroxylysyl pyridinoline, in the patient was 7.1 (controls: 0.20 ± 0.03; EDS VI: 5.99 ± 1.00[2]) and slightly increased (0.25 and 0.23) in the father and the mother, respectively, as also observed in other obligate heterozygotes[3]. Electrophoretic mobility of the α- and β-collagen chains of the various collagen types produced by cultured fibroblasts, performed as reported[9], was faster than normal which indicated that the lysyl residues were not hydroxylated and that there were no galactosyl and glucosyl-galactosyl residues attached. Mutation analysis, performed as described[10], on DNA of the patient showed a novel homozygous non-sense mutation in exon 12 of the PLOD1 gene at position 1302 of the cDNA sequence (c.1302 C>G) which leads to a premature stop at amino acid position 434 (p.Y434X; see also[11]). The parents were both heterozygous for the same mutation. Discussion Severe muscular hypotonia and kyphoscoliosis at birth, generalized joint laxity, and rupture of the ocular globes are considered major diagnostic criteria for EDS VI whereas tissue fragility including atrophic scarring, easy bruising, arterial rupture, Marfanoid habitus, microcornea, radiographically considerable osteopenia, and positive family history are classified as minor diagnostic criteria for the disease[1]. Since severe muscular hypotonia, joint laxity and kyphoscoliosis –which are three of the major criteria for EDS VI –are also common findings in neuromuscular disorders such as Ullrich muscular dystrophy and Bethlem myopathy, physicians are often misled to suspect them in EDS VI patients. Bethlem myopathy (OMIM 158810) and Ullrich muscular dystrophy (OMIM 254090) present with muscular weakness, hyperextensiblity of joints, and kyphoscoliosis and are caused by mutations in collagen type VI. Bethlem myopathy is an autosomal dominant disease characterized by proximal muscular weakness and joint contractures mainly involving the elbows, ankles and fingers[14] whereas Ullrich congenital muscular dystrophy is transmitted in an autosomal recessive mode and is characterized by severe muscular weakness of early onset so that children usually never develop the ability to walk because of contractures of proximal joints and hypermobility of distal joints. Scar formation due to poor healing is also evident in these patients[15,16]. The similarities of these conditions and EDS VI are striking and comprise severe hypotonia, kyphoscoliosis, hypermobility of joints and fragile skin causing scar formation[12]. Hence, extensive neuromuscular work-ups are performed usually in these patients yielding normal results before the diagnosis of EDS VI is considered[13]. The clue that could help physicians distinguish between EDS VI and neuromuscular disorders, specifically Ullrich muscular dystrophy, is the presence of microcornea, a possible history of rupture of arteries and of ocular globe, and osteopenia. The hypermobility of Ullrich dystrophy is limited to distal joints while in EDS VI all joints are involved. Our patient had remained undiagnosed until the age of 17, despite the presence of all four major, as well as all minor criteria (except for arterial rupture). Only recently this patient was referred to us for consultation. Multiple organ involvements, such as skin, muscle, bone and eye, made a diagnosis difficult. Indeed, a neuromuscular disorder was initially suspected based on the combination of severe hypotonia, delayed motor development, kyphoscoliosis and multiple dislocations. However, the presence of a positive history for ocular globe rupture, of velvety skin, microcornea and osteopenia –a pattern specific for EDS VI –helped us to reach the correct diagnosis without having to perform an invasive neuromuscular workup. Despite being considered a minor criterion, microcornea is a common finding in many of the previously reported EDS VI cases[2] as well as in our patient; osteopenia and recurrent joint dislocations are common findings, too, and were recorded in our patient as well. Ocular fragility is found in a minority of EDS VI individuals[2] and may lead to rupture of the globe after minor trauma as occurred in our patient. Vascular rupture, which is a life threatening complication, is absent in our patient at this age, and his parents were made aware of this potential complication later in life and were given advice in case of emergency for timely attending a hospital. Conclusion We suggest that physicians should consider EDS VI as one of the differential diagnoses in floppy infants and muscular weakness. The presence of positive findings such as microcornea, osteopenia, ocular rupture, arterial rupture is specific for EDS VI and can lead to the correct diagnosis. If EDS VI is suspected analysis of urinary pyridinolines should be performed next to confirm the diagnosis. The pyridinoline analysis is a sensitive, specific, robust, quick, and inexpensive method that reliably rules in or out EDS VI and helps to avoid an invasive neuromuscular work-up. In case of positive results further molecular testing is available to identify the disease causing mutation(s) and thus allows offering prenatal diagnosis for future pregnancies. To the best of our knowledge this is the first case report of EDS VI from Iran. Although the world-wide prevalence is estimated as low (~1 in 100.000 live births[4]) it might be more frequent in Iran because many of the reported and unreported (Giunta et al. in prep.) cases originate from Turkey, Greece, and Middle East[4] countries in which consanguineous marriages are frequent. With this case report we hope to increase the awareness of this connective tissue disorder and to help in the work-up of muscular weakness. Acknowledgement This work was supported by a grant from the Swiss National Science Foundation to B.S. (grant Nr.3200B0-109370/1). We thank Angelika Schwarze and Céline Bürer-Chambaz for skilful technical assistance. References
Copyright 2010 - Iran Journal of Pediatrics |

{kind=link}
{kind=link}