 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Iranian Journal of Pediatrics, Vol. 20, No. 2, Apri l-June, 2011, pp. 139-150 Molecular Diagnosis of Congenital Adrenal Hyperplasia in Iran: Focusing on CYP21A2 Gene Bahareh Rabbani1,2, PhD; Mahdieh Nejat3,PhD; Mohammad-Taghi Haghi Ashtiani2, MD; Mohammad-Taghi Akbari4, PhD; Ali Rabbani1,2, MD
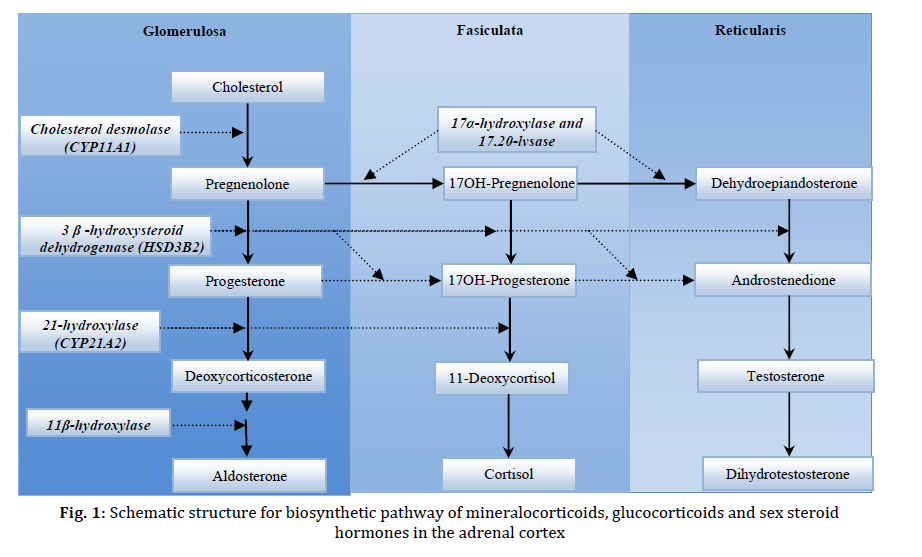
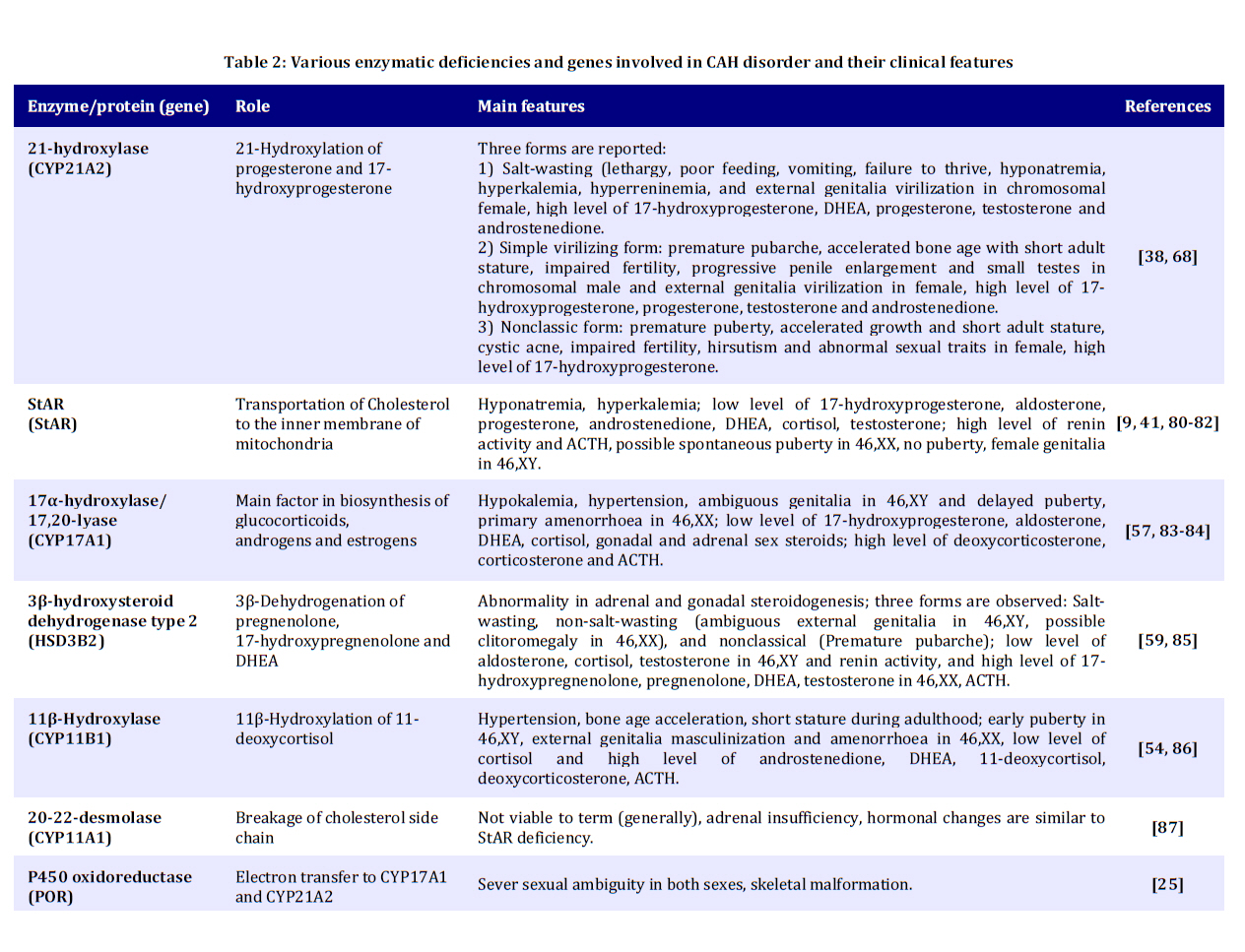
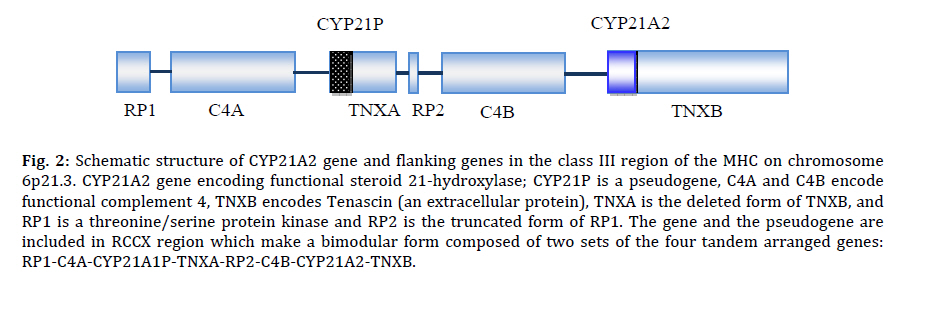
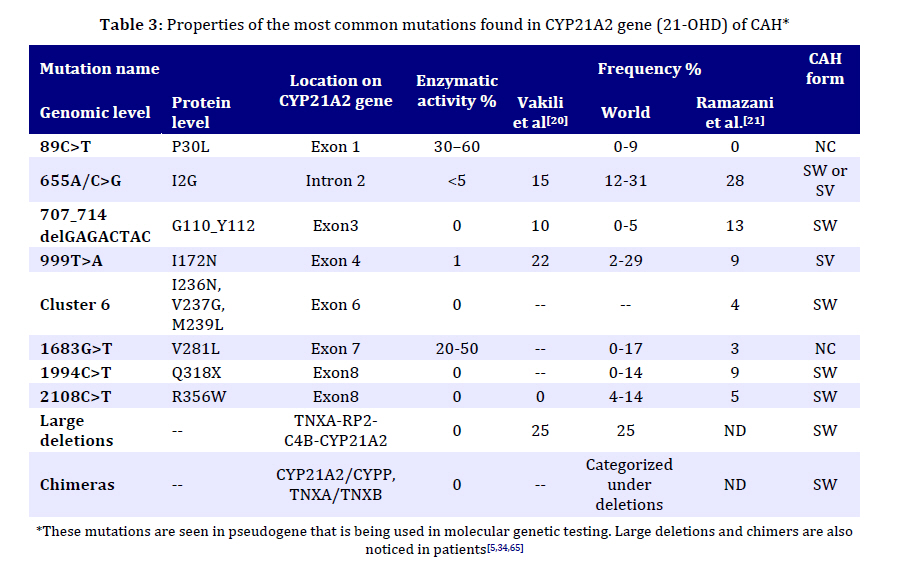
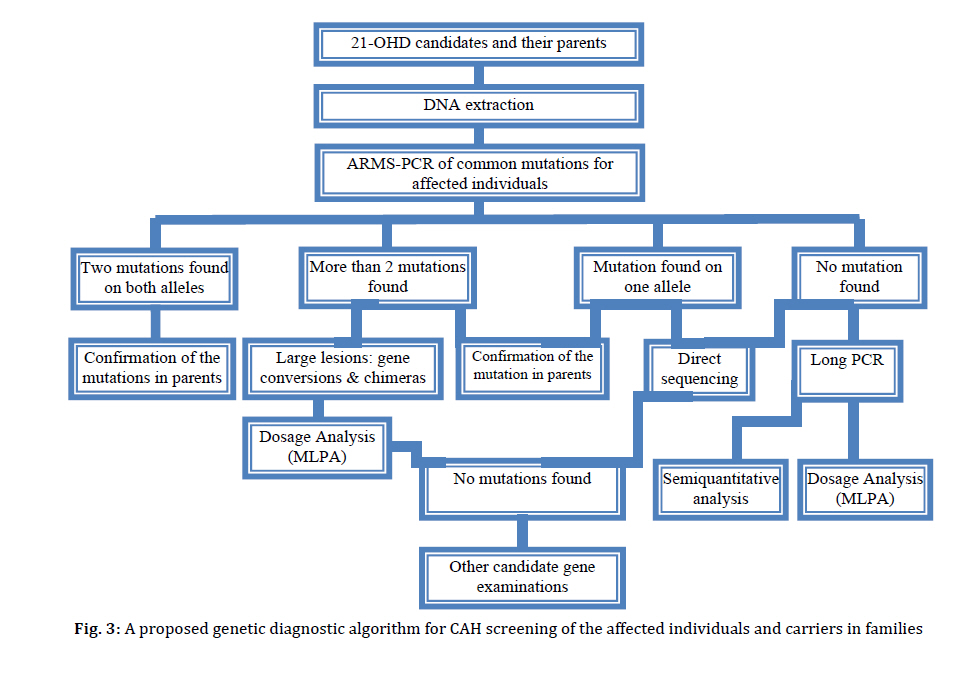
Received: Apr 21, 2010; Final Revision: Oct 23, 2010; Accepted: Dec 06, 2010 Code Number: pe11026 Abstract Congenital adrenal hyperplasia (CAH) is characterized by impaired biosynthesis of cortisol. 21-hydroxylase deficiency is the most common cause of CAH affecting 1 in 10000-15000 live births over the world. The frequency of the disorder is very high in Iran due to frequent consanguineous marriages. Although biochemical tests are used to confirm the clinical diagnosis, molecular methods could help to define accurate diagnosis of the genetic defect. Recent molecular approaches such as polymerase chain reaction based methods could be used to detect carriers and identify different genotypes of the affected individuals in Iran which may cause variable degrees of clinical expression of the condition. Molecular tests are also applied for prenatal diagnosis, and genetic counseling of the affected families. Here, we are willing to delineate mechanisms underlying the disease, genetic causes of CAH, genetic approaches being used in the country and recommendations for health care improvement on the basis of the molecular and clinical genetics to control and diminish such a high prevalent disorder in Iran. Also, the previous studies on CAH in Iran are gathered and a diagnostic algorithm for the genetic causes is proposed. Key Words: Congenital Adrenal Hyperplasia; 21 Hydroxylase Deficiency; Iranian population; Molecular Genetics Introduction Congenital adrenal hyperplasia (CAH) is one of the common endocrine disorders, caused by reduced or complete absence of enzymatic activities of steroid biosynthesis pathway (Fig. 1)[1,2]. Based on the phenotypic expression of the disorder, it is categorized into two major forms: severe or classic form, consists of salt-wasting and simple virilizing type, and late onset or non-classic form[3]. The variable clinical phenotypes depend on the reduced enzymatic activity due to different combination of gene mutations which may lead to mortality and morbidity. Steroids are widely produced in adrenal cortex which is classified into mineralocorticoids, glucocorticoids, or sex hormones. Aldosterone, end product of mineralocorticoids, synthesized in outer zona glomerulosa, affects the electrolyte balance. It helps to reabsorb sodium from kidney, colon and sweet glands. Cortisol, a glucocorticoid, is the main product of the fasciculata of adrenal cortex preserving carbohydrate metabolism, coping with stress, infection, and trauma. Sex hormones such as dehydroepiandrosterone (DHEA) and estradiol are secreted from zona fasciculata and reticularis. These steroids are regulated by a negative feedback of hypothalamus-anterior pituitary-adrenal axis[4]. Corticotropin releasing hormone (CRH) from hypothalamus stimulates adrenocorticotropoic hormone (ACTH) production. Low cortisol level has a significant feedback control on ACTH secretion[5]. Enzymatic defects in steroid pathway shunts the loop into other pathways, particularly androgen pathway which in turn leads to adrenal hyperplasia and over production of intermediate metabolites (Fig. 1)[4]. CAH mainly occurs due to mutations in genes including CYP21A2, StAR, CYP11A1, HSD3B2, CYP17A1 and CYP11B1 which encode the following enzymes: 21-hydroxylase (21-OH), steroidogenic acute regulatory protein (StAR) (lipoid adrenal hyperplasia), cholesterol 20-22 desmolase, 3β-hydroxysteroid dehydrogenase (3β-HSD), 17α-hydroxylase (17α-OH) and 11β-hydroxylase (11β-OH), respectively[6-11]. Basically the genes for the enzyme deficiencies are known, and mutational analysis is implemented in research and diagnostic laboratories around the world. There are many reports about CAH in Iran[12-19]. Clinical examination, paraclinical tests, HLA typing and karyotyping have been done in many of these studies. However, a few investigations have been reported on the genetic cause of Iranian patients[20,21]. The objective of this record is to gather information on the genetic basis of CAH, prenatal diagnosis, carrier detection, genetic counseling, and the genetic approaches which are being used in Iran; health improvement for patients and their families to reduce the physical, social, economical and psychological stress among the afflicted families is also one of the highlights of this country. Pediatricians should be aware of this kind of malformation and the genetic supports that could be carried out before the occurrence of any distress for the family and the community. Clinical manifestations Due to excess amount of androgen produced during gestation, virilization of the female fetuses in 21-OH deficiency and 11β-hydroxylase deficiency is noticed, causing female pseudohermaphroditism. Boys usually have no signs of androgen excess, although they generally show hyperpigmentation of genitalia, but may be diagnosed if being salt looser due to dehydration and shock[5,22]. Affected 46, XY boys with 17α-OH deficiency and lipoid hyperplasia have female-like external genitalia which is due to the impaired testosterone biosynthesis. Usually boys and girls affected with 3β-HSD deficiency have ambiguous genitalia. Females with 17α-OH deficiency remain sexually immature[22]. Symptoms including weight loss, poor appetite, dehydration, vomiting diarrhea, acidosis and failure to thrive may be attributed to the classic form of CAH for insufficient aldosterone maintenance as manifested in 21-OH deficiency, 3β-HSD deficiency, and lipoid hyperplasia. Patients with 11β-OH deficiency and 17α-OH deficiency may assign to hypertension due to high level of deoxycorticosterone (Table 1 and 2)[5,22,23]. Some patients only demonstrate signs of virilization, and do not show any signs of salt loosing despite showing pseudo-puberty with advanced bone maturation which are categorized under simple virilization[5]. Patients with mild, late onset, nonclassic form present with precocious puberty and accelerated growth rate in children and tall stature, infertility, acne, hirsutism, and oligomenorrhea in young women[5,24]. However, some asymptomatic forms may appear. Genetics The enzymatic deficiencies causing CAH mainly include any abnormality in production and activity of 21-OH, steroidogenic acute regulatory protein (StAR), lipoid adrenal hyperplasia, 3β-HSD, 17α-OH and 11β-OH, and a new form of CAH resulting from deficiencies in both 21-hydroxylase and 17α-OH/ 17,20-lyase activities were reported[25-27]. The main properties of different enzymatic defects causing CAH are presented in Table 2. The most significant cause of CAH is 21-OH deficiency, accounting for more than 90-95% of all cases[28]. The high frequency of 21-OH deficiency (347 of 433 CAH) has been also described among other forms of CAH in Iranian patients[14]. Thus, it is essential to have an overview on gene structure and mutations involving 21-OH deficiency. Classic form of 21-OH deficiency is the most prevalent variant of CAH, affecting 1 in 10000 to 1 in 15000 births[23,29,30]. The carrier frequency of CAH in the general population is 1 in 55[31]. The frequency of the non-classic form is difficult to determine owing to problems of ascertainment but it is determined to be approximately 1 in 100 live births[5]. All other forms of CAH occur at a frequency of 1/100000 in most populations[22]. The functional CYP21, CYP21A2 gene encoding 21-OH, consists of ten exons which is located on the short arm of chromosome 6 (6p21.3) in the class III region of the major histocompatibility complex (MHC)[32-34]. There is a non-functional pseudogene, CYP21A1[32], with 98% identity in the vicinity of CYP21A2 (nearly 30 kb apart from each other). These two genes together with the genes encoding complement proteins (C4A, C4B), tenascin (TNX A and B) which TNXB encodes an extracellular matrix protein[35,36], and serine/ threonine nuclear proteins (RP1 and RP2) are arranged in tandem manner as a bimodule (Fig. 2)[23,34,37,38]. It is generally accepted that the duplication of an ancient gene may lead to this configuration. There are many point mutations, small deletions, small insertions, splicing mutations, and gross deletions, duplications and chimeras found in CYP21A2 genes[34,39-41]. According to Human Gene Mutation Database (HGMD) there are more than 100 mutations found (www.hgmd.cf.ac.uk). The following common mutations in the CYP21A2 gene have been described in many populations accounting for three forth of the mutations: g.89C>T (p.P30L), g.655A/C>G (I2G), g.707_714delGAGACTAC (p.G110_Y112), g.999T>A (p.I172N), Cluster 6 (p.I236N, p.V237G, p.M239L), g.1683G>T (p.V281L), g.1994C>T (p.Q318X), and g.2108C>T (p.R356W). Different large deletions including all exonic parts of CYP21A2 account for 20-25% of 21-OH deficient cases in most populations[42]. Although these mutations have been extremely studied in many populations around the world[34,42-48], so far few studies have been published for Iranian population (Table 3)[20,21]. It is implied from Table 3 that there is an ethnic-specific manner mutation frequencies[48,49-51]. Thus, a comprehensive study on different populations around the country is essential to determine the mutation frequencies based on each ethnic cohort. CYP21A2 gene has a 98% identity to its pseudogene, CYP21P[32]. This structure is prone to recombination errors and unequal crossing-overs during meiosis resulting in CYP21A1P–CYP21A2 and TNXA–TNXB chimeras, large/short gene conversions, deletions and duplications[39,52]. Various combinations of gene deletions and conversions which may occur on the basis of different breakpoints complicate mutation analysis. Discriminating between different compound heterozygotes may also entangle the prediction of phenotypes. The deleterious alleles affect production and/or enzymatic activity of 21-OH and may show the phenotypic variability. The genetic background of the affected individual, modifier factors, other loci and association of different mutations may influence the phenotypes[38]. 11-OHD has been reported as the second cause of CAH in Iran accounting for 63 of 433 patients[14]. CYP11B1 gene located on chromosome 8q21 has nine exons which is 40 Kb apart from CYP11B2 gene for the aldosterone synthesis. CYP11B1 is amplified in a number of fragments and directly sequenced for mutation analysis[8,53,54]. CYP17A1 gene has been mapped on chromosome 10q24.3 encoding 8 exons which catalyze two enzymatic reactions, one 17α-hydroxylation and the other 17, 20-lyase reaction. For molecular testing direct sequencing is accomplished for all coding regions[6,55-57]. 6p21.3. CYP21A2 gene encoding functional steroid 21-hydroxylase; CYP21P is a pseudogene, C4A and C4B encode functional complement 4, TNXB encodes Tenascin (an extracellular protein), TNXA is the deleted form of TNXB, and RP1 is a threonine/serine protein kinase and RP2 is the truncated form of RP1. The gene and the pseudogene are included in RCCX region which make a bimodular form composed of two sets of the four tandem arranged genes: RP1-C4A-CYP21A1P-TNXA-RP2-C4B-CYP21A2-TNXB. 3β-HSD deficiency shows variable expression of both sexes. It is caused by mutations in HSD3B2 gene, located on chromosome 1p13.1, comprised of 4 exons. Molecular diagnosis of this gene is also carried out by sequencing of the polymerase chain reaction (PCR) products[11,58-60]. StAR gene is located on 8p11.2 which imports cholesterol to mitochondria. Instead cholesterol desmolase enzyme helps the cleavage of cholesterol side chain. The related gene is on chromosome 15q23-24[61-63]. Finally, it should be mentioned that other genes such as CYP11A1 and POR genes are also involved but have a rare frequency compared to other deficiencies. Diagnosis Biochemical CAH can be diagnosed on the basis of biochemical assessment of hormonal concentrations. Metabolites such as 17α-hydroxy-progesterone (17-OHP), androstenedione, dehyroepi-androsterone (DHEA), cortisol, testosterone, aldosterone, renin are assayed to determine the clinical variant of CAH which some may accumulate before the enzymatic block with the exception of lipoid adrenal hyperplasia that no steroid is produced in this form of disorder. Direct assessment of the enzymatic activity of 21-OH is impossible because CYP21A2 gene is expressed principally in the adrenal cortex. Based on accumulation of precursors after ACTH (cosyntropin) stimulation test[5], and comparing precursor:product ratios, the test is performed[5]. Normally, high concentration of serum 17-OHP is observed in classic 21-OH deficiency. For differentiating 21-OH deficiency from other steroidogenic enzymatic defects ACTH stimulation test is performed[23,30]. A profile of steroid metabolites is needed to evaluate the clinical cause of the disease. However, we cannot use this method for accurate diagnosis of carriers, and asymptomatic patients. Determination of genotype in affected individual also cannot be done using biochemical test. Molecular As consequence of the pseudogene existence, the genetic diagnosis of 21-OH deficiency and interpretation of the results (especially chimeric genes, gene conversions and duplications) are more difficult than other single gene disorders. Molecular tests for diagnosis of genetic causes of CAH have been set up since many years ego. The eight common mutations have been studied in 85 Iranian patients[20,21]. However, a proposed algorithm presented in Fig. 3 shows a diagnostic protocol for 21-OH deficient patients which was investigated for more than 50 affected families and also is being performed at present in Pediatric Center of Excellence, Children’s Medical Center. Briefly, the following procedure was performed for the genetic testing of CAH affected families. Clinically examined affected individuals were selected for genetic testing. 21-OH deficient candidates were enrolled for the molecular analysis. Informed consent was obtained from affected families. DNA extraction was implemented from affected individuals and their parents (and siblings if available). Then, common mutations of CYP21A2 gene were investigated by a PCR based technique, amplification refractory mutation system (ARMS) at first step. PCR conditions for these mutations have been described previously[64,65]. These techniques could differentiate between carriers and normal individuals, indicating heterozygotes and homozygotes. If no mutation or just one mutation was found, sequencing (ABI 3730, Applied BioSystems, US) of all exonic regions of the CYP21A2 gene[66] was carried out to detect rare and novel point mutations[67]. If a large deletion (partial or complete gene conversion, chimers) and duplication were suggested, multiplex ligation probe amplification (MLPA) was performed to confirm it with the use of capillary electrophoresis (ABI 3010 Applied BioSystems, US) (Fig. 3). MLPA kits (MRC-Holland) are available for dosage screening purposes[68,69]. Recently, PCR and real time PCR have been applied for diagnosis of large deletions[70-72]. We found common mutations of CYP21A2 gene. Gene duplication, deletion [73], large gene conversion and rare mutations were also found with the help of MLPA and sequencing techniques (Our unpublished data). We are analyzing other involved genes such as HSD3B2 and StAR genes in more patients. Genetic Counseling Individuals who have inherited a mutation are at risk of transmitting it to their offspring. The risk of first cousins for autosomal recessive disorders is 6-8%. An appropriate genetic counseling can be helpful and beneficial for families having affected individuals. Genetic counseling provided for the affected families is dependent on the genotypes diagnosed by molecular techniques. All reported CAH mutations are inherited recessively. Thus, the recurrence risk of 25% is recognized for parents with a pervious affected child, although de novo mutations in the CYP21A2 gene have a significant role in introducing new alleles to populations; their frequency is 1–2%[74]. Various ethnic groups with different cultures including Persian, Azari, Kurd, Gilaki, Lur, Turkmen, Arab, etc. exist in Iran. Because of high prevalence of some mutations in different groups in other genetic diseases such as congenital hearing loss, ethnicity is considered[75,76]. Carrier screening is essential for any population with high frequency of intra group marriages and at risk families. Hence, molecular diagnosis of the mutations is required for carrier detection, genetic counseling and prenatal diagnosis. Prenatal Diagnosis Nowadays, one of the important parts of pregnancy care is prenatal diagnosis. This process is applied to determine the disease of a fetus before birth. One of the aims of prenatal diagnosis is to reduce genital ambiguity and subsequent problems each individual is dealing with in CAH. Luckily, 21-OHD is a preventable disease in females with ambiguous genitalia when it is determined by prenatal diagnosis methods[77]. Many approaches such as biochemical diagnosis are being applied for prenatal diagnosis of affected fetuses since 1965 by Jeffcoate and colleagues. Recently, molecular genetic techniques are increasingly used to genotype the CYP21A2 gene from extracted DNA of fetus. Chorionic villus sampling (CVS) and amniocentesis are the main sources for extracting fetal DNA at 8th-9th and 12th-13th weeks of gestation, respectively. New studies point out that for affected or high risk families, dexamethasone administration is started at the 8th week of gestation. If karyotyping or DNA analysis shows that the fetus is a male, or the fetal DNA is unaffected for female, dexamethasone is stopped; if not, it is continued to term[77-79]. Conclusion CAH is one of the main pediatric referent, due to genital ambiguity and mortality of neonates as a result of dehydration and shock. Molecular diagnosis can be made for high risk families, before spending any unnecessary and excessive expenses, which also cope with socioeconomical, and psychological problems, thus this would avoid genitoplasy for the affected individual. However, mis-diagnosis of asymptomatic nonclassic forms and heterozygotes has many undesirable consequences that show up later in life. Therefore, carrier detection and genetic counseling of the families having an affected child would diminish the complications for the families. Bearing in mind, genetic testing would assist accurate diagnosis of the affected individuals, and identify their genotypes; therefore, it could be used in genetic counseling to reduce genital virilization of fetus and other difficulties the child and the family would deal with. Thus, a health care policy program based on molecular genetics draws the only essential way to decrease the incidence of CAH in our country. Acknowledgment We would like to appreciate the families involved in this report. We wish to thank the staffs of Molecular Genetic Lab, Children's Medical Center Hospital, and Pediatrics Center of Excellence for helping in biochemical and genetic tests. These tests were supported by grants of Growth and Development Research Center, and Endocrine and Metabolism Research center of Tehran University of Medical Sciences. Grant supporting number 7383 of Tehran University of Medical Sciences. Conflict of Interest: None References
Copyright 2011 - Iran Journal of Pediatrics The following images related to this document are available:Photo images[pe11026t1.jpg] [pe11026f3.jpg] [pe11026f2.jpg] [pe11026f1.jpg] [pe11026t3.jpg] [pe11026t2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}