 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
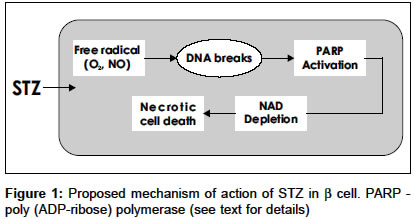
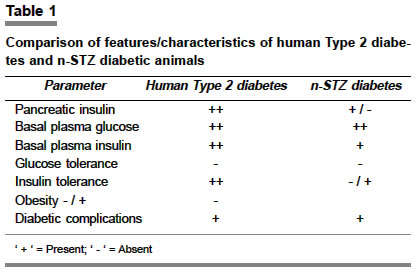
Indian Journal of Pharmacology, Vol. 36, No. 4, August, 2004, pp. 217-221 Education Forum Neonatal streptozotocin-induced rat model of Type 2 diabetes mellitus: A glance Arulmozhi DK, Veeranjaneyulu A, Bodhankar SL Department of Pharmacology, Bharati Vidyapeeth Deemed University, Poona College of Pharmacy, Erandwane, Pune - 411 038 Code Number: ph04073 ABSTRACT Diabetes mellitus is a group of syndromes characterized by hyperglycemia, altered metabolism of lipids, carbohydrates and protein and an increased risk of complication of vascular diseases. Type 2 diabetes mellitus is characterized by derangement of insulin secretion and an inability of the peripheral tissues to respond to insulin. In spite of the availability of many animal models for Type 2 diabetes mellitus including genetic and chemically induced, none of them simulate human Type 2 diabetes mellitus. An attempt has been made in the present review, to evaluate the neonatal streptozotocin-induced rat (n-STZ rats) model of Type 2 diabetes mellitus, for its potential advantages as a suitable model over the others. The n-STZ model (with alteration of dose and day of STZ injection) exhibits various stages of Type 2 diabetes mellitus such as impaired glucose tolerance, and mild, moderate and severe glycemia. The cells in n-STZ rats bear a resemblance to insulin secretory characteristics found in patients with Type 2 diabetes mellitus. Thus the n-STZ model can be considered as one of the suitable animal models of Type 2 diabetes mellitus. Keywords: Beta cells, NIDDM, insulin, hyperglycemia Diabetes mellitus, a metabolic disorder, is characterized by hyperglycemia, altered metabolism of lipids, carbohydrates and proteins with an increased risk of complication of vascular diseases.[1],[2] The minimum defining characteristic feature to identify diabetes mellitus is chronic and substantiated elevation of circulating glucose concentration.[2],[3] Diabetes mellitus may present as a relatively sudden, potentially lethal metabolic catastrophe or it can be associated with few if any, symptoms or signs and may escape detection for many years. These extremes of clinical manifestations constitute the basis for subdividing diabetes mellitus into the insulin dependent (IDDM) and the non insulin dependent (NIDDM) types. Type 1 diabetes mellitus results from a severe absolute lack of insulin, caused by reduction in the b cell mass. The three interlocking mechanisms responsible for the islet cell destruction are genetic susceptibility, acute auto immunity and environmental insult.[1],[2] Type 2 diabetes mellitus identifies patients who do not require insulin treatment to remain alive. The two metabolic defects characterizing Type 2 diabetes mellitus are 1. derangement of insulin secretion that is delayed or is insufficient relative to glucose load and 2. inability of peripheral tissues to respond to insulin - called insulin resistance. Though the primacy of one or the other of these defects is a matter of debate, most of the patients have relative or absolute deficiency of insulin.[1] Animal models of Type 2 diabetes mellitus 1. Genetic and spontaneous models Sand rat (Psammomys obesus) is a model of nutritionally-induced Type 2 diabetes mellitus. Psammomys is prone to developing hyperinsulinemia, hyperglycemia and obesity when transferred to a high-energy diet, also primary insulin resistance is a species characterization of Psammomys. However, the potential to become diabetic decreases with age. In Psammomys of ages 1-12 months, maintained on a low-energy diet from weaning and transferred at different ages to a high-energy diet,[3] the sensitivity to the development of diabetes mellitus increases from weaning to a peak of about 5 months of age and decreases thereafter. The fatty Zucker (Zucker diabetic fatty (ZDF) rat has been valued as a model of obesity, as the characteristics of the model are described as hyperglycemia, early hyperinsulinemia, fasting hyperglycemia, abnormal glucose tolerance, hyperlipidemia, mild hypertension. The ZDF rat carries the Lepfaor Ob-Rfa mutation, which is typically referred to as just fa.[4] The JCR: LA-Cp rat is a rodent model possessing insulin resistance, obesity, hypertriglyceridemia, including end stage cardiovascular diseases. This genetic model possesses the autosomal recessive gene called cp (corpulent), however, the elements that lead to the cardiovascular disease in this model are unknown.[5] Metabolic syndrome X consists of insulin resistance as a primary defect associated with compensatory hyperinsulinemia, impaired glucose tolerance, dyslipidemia and hypertension.[6] The obese spontaneously hypertensive rat (SHORB) is considered as a model of metabolic syndrome X. The obese phenotype results from a mutation of leptin receptor designated fak. SHORB is a useful model to understand the interaction of the various metabolic abnormalities that make up syndrome X.[7] The obesity mutations in the mouse ob (obese) and db (diabetic) are mutation in the leptin structural gene (ob) and mutation in the leptin receptor gene (db). The obese hyperglycemic syndrome displayed by these mice shows various similarities to the metabolic abnormalities present in syndrome X and Type 2 diabetes mellitus. Insulin resistance, inappropriate hyperglycemia, impaired glucose tolerance as well as increased insulin secretion finally leading to b cell exhaustion are seen in these models. However age, gender, and maintenance conditions are reported to affect the phenotype of these mice.[8] The spontaneously diabetic KK mice are reported to have moderate obesity, polyphagia, polyurea, persitent glycosuria, glucose intolerance, moderate hyperglycemia, hyperlipidemia, insulin resistance of peripheral tissues and hyperinsulinemia. The diabetic characteristics of KK mice and the variant KKAy are reverted to normal after 40 weeks of age.[9] The OLETF (Otsuka - Long - Evans - Tokoshima - Fatty) rat is a spontaneously diabetic rat with characteristic features of late onset hyperglycemia (after 18 weeks of age), a chronic disease state, increased urinary protein excretion, higher glomerular filtratin rate, increased kidney weight etc. Thus the clinical and pathological features of the disease state in OLTEF rats resemble those of human renal complications in human Type 2 diabetes mellitus.[10] The Cohen diabetic rat is an exceptional, genetically derived, diet-induced Type 2 diabetes mellitus model that expresses genetic susceptibility to a carbohydrate-rich diet, a central feature of Type 2 diabetes mellitus in humans. A major drawback of this model is that it had never been systematically characterized in terms of phenotype or genotype, resulting in only limited recognition of its value and potential contribution to diabetes research.[11] 2. Experimentally-induced models Type 2 diabetes mellitus induced by streptozotocin (STZ): Streptozotocin is a deoxy-s [((methyl-nitrosoamino) carbonyl)-amino]-D gluco pyranose molecule that produces a selective toxic effect on b cells and induces diabetes mellitus in most laboratory animals.[12],[13] High doses of b cell toxins like streptozotocin and alloxan induce insulin deficiency and Type 1 diabetes mellitus with ketosis. However, doses calculated to cause a partial destruction of b cell mass can be used to produce a mild insulin deficient state of Type 2 diabetes mellitus, without a tendency to cause ketosis.[14] The dosage is difficult to judge to create stable Type 2 diabetes mellitus without either gradual recovery or deterioration into Type 1 diabetes mellitus. Streptozotocin is preferred because it has more specific b cell cytotoxicity, but the sensitivity of this agent varies with species, strain, sex and nutritional state and there are batch differences in activity.[15] The Okamoto rat model is a model for b cell damage and the prevention of the damage was based on results obtained in rodents with STZ and alloxan-induced diabetes mellitus.[16] Recently, a newer rat model of STZ diabetes mellitus has been reported using suitable doses of nicotinamide against the b-cytotoxic effect of STZ. The diabetic syndrome developed in adult rats by injecting nicotinamide and STZ, reported to exhibit comparable features with human Type 2 diabetes mellitus. The literature reveals that sulfonylureas and sodium tungstate are effective in this model. The nicotinamide-STZ model is further validated in mini pigs, in which the glucagon-like peptide-1 derivative NN 2211 has been reported to be effective. However, the optimal dose of nicotinamide to cause a stable hyperglycemia is a major concern with this model.[17],[18],[19],[20],[21] Suggested mechanism of STZ: Although the exact mechanism of its toxicity is still a matter of debate, one proposed site of action of STZ is at nuclear DNA. During the decomposition of STZ, highly reactive carbonium ions are formed, which cause alkylation of DNA bases[13] and also STZ may damage the b cell membrane and break the DNA strand which leads to the activation of poly (ADP-ribose) synthetase and NAD depletion, which ultimately leads to cell death [Figure - 1].[14],[15] Neonatal STZ-induced rat (n-STZ) model of Type 2 diabetes mellitus: The diabetic syndrome in this model is generated by injecting Wistar rats on the day of their birth (n0=birth) intravenously (sapheneous vein) or intraperitoneally with 100 mg/kg of STZ.[15] Also, the n-STZ rat model is developed by varying the day of the STZ injection after the birth, such as 2nd day or 5th day of the birth, and these are alternatively called n2-STZ and n5-STZ models respectively.[22],[23] The n0-STZ model: The rats treated with STZ on the day of birth, exhibit insulin deficient acute diabetes mellitus 3-5 days after birth. They showed high plasma glucose and about 93% decrease in plasma insulin and high plasma glucagon content. The hyperglycemia observed in the neonates following STZ is only transient.[24],[25] It is reported that the neonatal rats are resistant to STZ.[15],[25],[26] The plasma glucose and insulin values are no longer significantly different from those of controls. It was found that only by 8 weeks of age and thereafter n0-STZ rats showed mild hyperglycemia.[23],[24] Other n-STZ models: As discussed, the n2-STZ and n5-STZ models are developed by 80 mg/kg, i.p. STZ injection on the 2nd day and 5th day of the birth respectively.[24],[25] An interesting variant of the model has been reported: Sprague-Dawley pups were injected intraperitoneally on the 2nd day after birth with 90 mg/kg STZ and on 1.5 days after birth with 120 mg/kg STZ.[23],[24] By 6 weeks of age these animals showed basal hyperglycemia and abnormal glucose tolerance.[26],[27] The n0-STZ and n2-STZ Wistar models are found almost similar with respect to growth, basal plasma glucose, insulin levels, lack of insulin release in response to glucose in vivo, glucose intolerance and depletion of pancreatic insulin stores.[27],[28],[29] The n5-STZ Wistar model showed an unaltered basal hyperglycemia, glucose intolerance, raised glycosylated hemoglobin, a strong reduction of the pancreatic insulin stores, decreased (50%) basal insulin levels and lack of plasma insulin response to glucose in vivo. The development and progression of hyperglycemia found in the n5-STZ Wistar models demonstrated many similarities to those of the n2-STZ Sprague-Dawley model.[26],[27] The b cell function in n-STZ models Quantitative and qualitative defects in insulin secretion in Type 2 diabetes mellitus: Type 2 diabetic subjects display more subtle changes in the dynamics of insulin secretion, such as blunting of the first phase insulin secretion and disruption of the insulin secretory pulses.[29] The first phase is a very brief surge of insulin that follows an acute secretagogue challenge such as an intravenous glucose bolus. It peaks after 2-4 min and dissipates within 6-10 min. If the challenge is sustained, the prolonged second phase starts; the second phase supervenes and lasts until the glucose is cleared. The first phase is a more efficient signal than the second, both in enhancing glucose clearance and priming the liver to shut down glucose production.[28],[29] Glucose regulates b cell function in two ways. It produces a direct release of insulin as a result of enhancement of the concentrations and its pre-stimulus level modulates the response to the islet secretagogues.[30] It is also suggested that the first phase response to glucose is specifically abolished in Type 2 diabetes mellitus. The second phase insulin secretion is also attenuated. It is not clear which defect has a greater impact on glycemia. Insulin, like other hormones, is secreted in pulses and this appears to be a fundamental signal for hormone signaling.[30],[31] In normal subjects under fasting conditions, insulin release occurs in regular pulses with a periodicity of about 13 min.[32] But in subjects with Type 2 diabetes mellitus insulin secretory profiles are more chaotic[32],[33] and also the regular 13 min pulses are absent.[1] Defects in n-STZ models: Adult n0-STZ rats are characterized by a low insulin release in vivo in response to glucose or amino acids.[15] In the insulin secretion studies of the 10-16 week-old n0-STZ rats, there was a complete loss of b cell sensitivity to glucose.[23] The insulin pulse amplitude was also affected by mild to moderate b cell damage induced by STZ.[26],[27] The impairment of glucose-induced insulin release in n-STZ rat is clearly related to a defect in oxidative glycolysis. This leads to a severe decrease in the mitochondrial oxidative catabolism of glucose-derived pyruvate. It coincides with a lower ATP/ADP ratio in simulated islets and their subsequent alteration of ionic events rightly coupled to the fuel function of the hexose in the islet cells.[27] It has been found that the n-STZ rats exhibited an increased amylin-insulin molar ratio. This has been identified as a major component of amyloid deposits in the pancreatic islets of patients with Type 2 diabetes mellitus.[32],[34] Defects in exocrine pancreas in n-STZ rats: After STZ injection, the pups showed an increase in pancreatic weight and pancreatic protein and DNA content at Day 16 as compared to the control groups, but not thereafter. After Day 16 of the STZ treatment, the n-STZ pups showed a transient increase in their pancreatic lipase and trypsinogen concentration in contrast to pancreatic amylase. The amylase levels were found lowered in STZ pups from Day 18 to Day 42. But the difference between groups was statistically significant only from Days 18 to 24. Insulin supplementation of STZ pups for fewer days in the neonatal period restored the pancreatic amylase concentration to the control level at Days 18-20. This indicated that acute STZ administration at birth to neonates affects subsequent exocrine pancreas development, particularly that of amylase while exogenous insulin attenuated the effect.[33] b cell regeneration in n-STZ rats: It has been reported that after the n0-STZ injection, from the postnatal Day 4 onwards, signs of regeneration are apparent, in that numerous insulin positive cells are found throughout the acinar parenchyma and within the duct epithelium, but in 4-month-old animals the regeneration process was incomplete.[15],[23],[26] The timing of the STZ injection is the critical factor for the efficiency of the regeneration process, which coincides with the normal development of islet cell mass in the rat.[34] The n5-STZ rats showed lack of significant re-accumulation of insulin in the pancreas after 2 weeks following the b cell insult, but some degree of recovery in the insulin stores was found in the pancreas of the n2-STZ and n0-STZ rats. It has been proved that there is some capacity of b cell regeneration in the neonatal rat pancreas (which is lacking in the adult rodents) [35] and the capacity of the b cell regeneration in the Wistar strain decreases quickly during the first postnatal week and thereafter it is no longer significant.[23],[24],[25],[26] It is also explained that the regeneration of the b cells in the Sprague-Dawley neonates is less efficient than in the Wistar strain. The recovery from diabetes mellitus in the Sprague-Dawley n2-STZ model is due to the partial replenishment of the b cell mass from the replication of the existing b cells, rather than neogenesis from undifferentiated precursors.[34],[35] n-STZ Type 2 diabetes mellitus: Is it reversible? It has been reported that after a STZ-induced injury, the surviving b cells are able to maintain most of their metabolic functions but fail to maintain an adequate insulin production.[23],[24],[25] When the n0-STZ model is included for insulin therapy, the basal glucose levels are decreased, but the pancreatic insulin stores are not affected by the insulin therapy, this needs a minimum of 5 days of insulin therapy.[36] It is hypothesized that the chronic hyperglycemia-hypoinsulinemia in the n0-STZ rat causes abnormal glucose influence on the glucose and arginine-stimulated insulin release.[36] In the Sprague-Dawley n2-STZ model the defects of glucose-induced insulin secretion were not restored by insulin treatment.[26],[27] It is noteworthy to recognize that the efforts to restore glucose-induced first phase insulin release in humans with Type 2 diabetes mellitus have not been totally successful.[26],[28] Interestingly, insulin therapy corrected the insulin response to glucose in the n0-STZ rat model, supporting the view that mild hyperglycemia and hypoinsulinemia contribute to the defective insulin response to glucose.[22] Insulin resistance in n-STZ models In contrast to the above findings, in 8-week-old n0-STZ female rats, it was shown that hepatic glucose production measured in the basal state was higher in the diabetes mellitus models than in the controls, despite similar peripheral insulin levels in both groups.[22] It is found that in white and brown adipose tissues, an increased responsiveness to insulin action is detected when comparing diabetic females to control females and insulin action was found normal in the skeletal muscles and diaphragm of the same adult females. The observations of the hormonal insulin action in the liver and white and brown adipose tissues indicated that glucose is preferentially channeled towards the liver and adipose tissue in n0-STZ females. The clamp studies in the n5-STZ model showed that the glucose utilization by the whole body mass induced by hyperinsulinemia was significantly reduced and the heptatic glucose production rate was less efficiently suppressed by submaximal or maximal insulin levels, which indicated that the insulin resistance is present in vivo at the level of the peripheral tissues and the liver.[36],[37],[38] As discussed, this confirmed that when b cell insult is the primary factor responsible for the emergence of moderate to severe hyperglycemia in rats, insulin resistance can develop secondarily[39],[40] and a certain degree of insulin deficiency is necessary to induce insulin resistance.[41],[42],[43] Is n-STZ rat a better model of Type 2 diabetes mellitus? By altering the dose and the day of the STZ injection, the n-STZ models exhibit various stages of Type 2 diabetes mellitus, such as impaired glucose tolerance, mild, moderate and severe hyperglycemia. The n-STZ rats exhibit slightly lowered plasma insulin levels, slightly elevated plasma glucose levels and lowered pancreatic insulin content.[40],[44] The b cells in the n-STZ rats bear a resemblance to the insulin secretory characteristics found in Type 2 diabetic patients [Table - 1].[22],[45],[46],[47],[48] The pattern of insulin release found in the n0-STZ and n2-STZ rats is qualitatively similar to that of the GK (Goto-Kakizaki) rat, which is a genetically diabetic non-obese model of human diabetes mellitus.[47] CONCLUSION It is understood that no animal model is identical to any human syndrome; none of the available animal models of Type 2 diabetes mellitus exactly simulate the human Type 2 diabetes mellitus. However, n-STZ rat models have several advantages over the other models as described above and is considered to be one of the suitable experimental animal models of Type 2 diabetes mellitus. ACKNOWLEDGEMENTS The authors wish to thank Dr. S. K. Arora, President, Lupin Research Park, Pune and Drs. S. S. Kadam &. K. R. Mahadik, Bharati Vidyapeeth Deemed University, Poona College of Pharmacy, Pune for their constant encouragement. REFERENCES
Copyright 2004 - Indian Journal of Pharmacology The following images related to this document are available:Photo images[ph04073f1.jpg] [ph04073t1.jpg] |
| |||||||||

{kind=link}
{kind=link}