
|
Indian Journal of Pharmacology
Medknow Publications on behalf of Indian Pharmacological Society
ISSN: 0253-7613 EISSN: 1998-3751
Vol. 37, Num. 6, 2005, pp. 348-357
|
Indian Journal of Pharmacology, Vol. 37, No. 6, November-December, 2005, pp. 348-357
Methods
Experimental animal models to induce cardiac arrhythmias
Bhatt LK, Nandakumar K, Bodhankar SL
Department of Pharmacology, Poona College of Pharmacy,
Bharati Vidyapeeth Deemed University, Erandwane,Pune - 411 038, India
Correspondence Address: S.L. Bodhankar, Department of Pharmacology, Poona
College
of
Pharmacy,
Bharati Vidyapeeth Deemed University, Erandwane,Pune - 411 038, India, E-mail:
sbodh@yahoo.com
Code Number: ph05095
Abstract Cardiac arrhythmias are of different types based on their mechanism and origin. The information gathered from animal studies has been instrumental in devising diagnostic and therapeutic strategies; so different animal models are needed for different types of arrhythmias. The origin and mechanism underlying clinical arrhythmias are of considerable significance, since knowledge of these processes may provide a basis for successful therapy. Various animal models that encompass different types of arrhythmias are reviewed. This review classifies various experimental models according to their origin, which are mainly supraventricular and ventricular. Also included are various transgenic animal models for arrhythmias.
Keywords: Atrial fibrillation, atrial flutter, re-entrant arrhythmia, ventricular fibrillation
Introduction
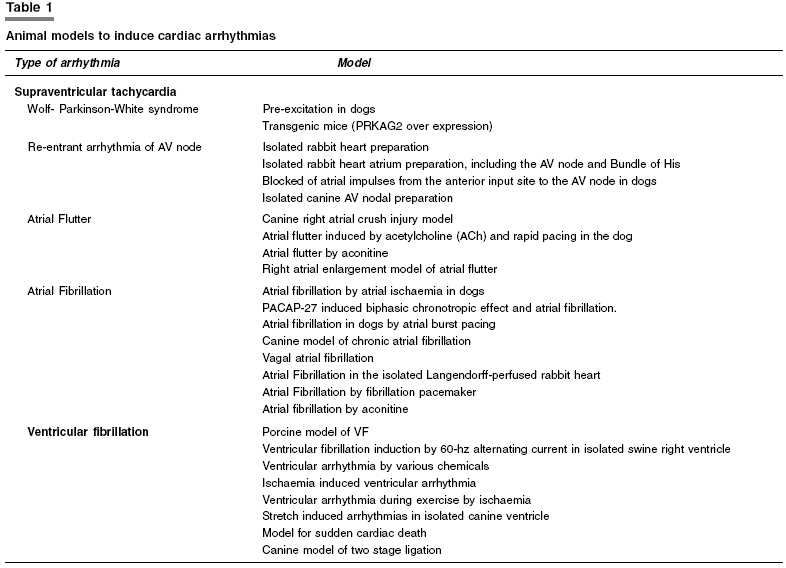
Arrhythmias are disorders of heart rhythm. They are due to abnormalities in impulse generation, impulse conduction, or a combination of both. Abnormalities of impulse generation include abnormalities of automaticity and early or delayed after depolarization with triggered activity. Abnormalities of impulse propagation include conduction block and re-entry of the cardiac impulse. Combination of abnormalities of impulse formation and propagation can produce complex arrhythmias.[1] In any arrhythmia, it is useful to know which cardiac tissue participates, the ionic mechanisms and structural abnormalities that promote it. Supraventricular and ventricular arrhythmias differ in origin, ECG changes and clinical manifestations, based on which one must be able to distinguish between supraventricular from ventricular arrhythmias. The mechanism underlying clinical cardiac arrhythmias are of considerable significance and it is unfortunate that these arrhythmias are not easily studied in clinical situations. Now a days, sophisticated electro-physiological techniques are available to study cardiac pathophysiology, both in vivo and in vitro. These techniques have enabled to study the underlying mechanisms of arrhythmias and conduction disturbances in both experimental models and in patients. Although our knowledge of the mechanisms of arrhythmias and conduction disturbances has greatly increased, much remains to be explored. Various animal models [Table
- 1] have been developed for supraventricular as well as ventricular tachycardia to understand the basic cause, origin, possible mechanisms, manifestations and for development of new therapeutic strategies. Supraventricular tachycardia in an animal model closely resembles the clinical features observed in the patients. But ventricular models are fraught with problems since they cannot be studied in human patients because of the unpredictable occurrence in situations, where electrophysiological changes may develop within minutes. Besides this, many other factors determine whether, and if so how often-ventricular arrhythmias occur in the setting of acute ischaemia and/or a chronic myocardial infarction. In experimental models, usually only a single factor is taken into account. Though, an animal is not the same as a human patient, arrhythmogenic mechanisms derived from animal experiments have tremendously helped us to diagnose and adapt therapeutic strategies.
The therapeutic strategies to treat cardiac arrhythmias include pharmacologic
approaches, ablation of specific foci involved in arrhythmogenesis, antiarrhythmic
surgical approaches and implantable devices designed to respond to tachyarrhythmic
events or to prevent symptomatic bradyarrhythmias. The antiarrhythmic
drugs may be classified according to the modified Vaughan Williams[2] system,
which categorizes them on the basis of electropharmacologic and electrophysiological
properties. Drugs having class-I action possess local anesthetic or membrane
stabilizing activity. Their predominant action is to block the fast inward
sodium channel. This produces a decrease in the maximum depolarization
rate of the action potential (Phase 0) and slows intracardiac conduction.
These agents can be further subclassified as class Ia, Ib or Ic on the
basis of their effects on specific aspects of intracardiac conduction and
refractoriness. Class-II drugs block β-receptor and thus reduce heart
rate, decrease intracellular Ca+ 2sub overload and inhibit
after depolarization-mediated automaticity. Class-III antiarrhythmic agents
prolong action potential duration, presumably through blockade of K+ channels.
Class IV antiarrhythmic agents inhibit the slow calcium influx during the
plateau of the action potential through Ca2+ channel
blockade.
Animal models to induce cardiac arrhythmia
Supraventricular tachycardia
Wolf-Parkinson-White syndrome (WPW syndrome)
The WPW syndrome, an electrocardiographic pre-excitation pattern, is associated in a fairly large percentage of cases[3] with
attacks of supraventricular tachycardia. At present, all the electrophysiological
characteristics of accessory atrioventric′′ular connections
and their role in causing re-entrant tachycardia have been obtained from
studies on human patients.[4] Boineau and Moore described pre-excitation in dogs and studied propagation of activation across an accessory atrioventricular connection in types A and B pre-excitation. In type A, the effective refractory period of the accessory pathway exceeds that of the normal AV nodal His-Purkinje pathway. Therefore, a premature atrial impulse may get blocked at the accessory pathway and conducted anterogradely down the normal pathway. Ultimately, entering the accessory pathway in the retrograde direction and re-entering the atrium to establish a circus movement tachycardia referred to as orthodromic. In type B, a shorter refractory period in the anomalous pathway and then retrograde invasion of the normal pathway, with antegrade conduction down the anomalous pathway and then retrograde invasion of the normal AV nodal pathway to establish an antidromic tachycardia. In type A, the delta wave and QRS complex are predominantly upright in the precordial leads. The dominant R wave in lead V1 may be misinterpreted as right bundle branch block. In type B, the delta wave and QRS complex are predominantly negative in leads V1 and V2 and positive in the other precordial leads, resembling left bundle branch block. In this study, they observed that atrial fibrillation induced in the dog caused ventricular fibrillation as well because the accessory pathway had a short refractory period and conducted many impulses, which otherwise would have been blocked in the AV node.[5]
Human mutations in PRKAG2, the gene encoding the γ2 subunit of AMP-activated protein kinase (AMPK), cause cardiomyopathy, characterized by ventricular hypertrophy, WPW syndrome and progressive conduction system disease.[6],[7] Michael et al . developed transgenic mice over-expressing the PRKAG2 cDNA with or without a missense N4881 human mutation. Transgenic mutant mice showed elevated AMP-activated protein kinase activity, accumulated large amount of cardiac glycogen, developed dramatic left ventricular hypertrophy and exhibited ventricular preexcitation and sinus node dysfunction.[8]
Drugs, which can be screened through these models, are adenosine type drugs and class Ia drugs for acute therapy. For chronic therapy class I as well as class III drugs can be screened. Classes II and IV (phenylalkylamine and benzothiazepine like) drugs, which can cause AV nodal block, can also be screened for acute therapy.
Re-entrant arrhythmia of AV node
Paroxysmal supraventricular tachycardia (PSVT) due to AV nodal re-entry
is the most common form of supraventricular arrhythmia. The underlying
pathophysiology in AV nodal reentry is the presence of dual AV nodal
pathways.[9] The AV node in
patients with dual-pathway physiology behaves as though there are two
types of conduction pathways in the AV node, one capable of faster conduction,
which usually has a longer refractory period, and the other more slowly
conducting and having a shorter refractory period.[10]
The pioneering clinical studies of the 1980s allowing successful surgical
treatment or catheter ablation[11],[12] of
the arrhythmia, all quoted the microelectrode studies on the isolated
rabbit heart preparations that provided insight into arrhythmia mechanisms
on a cellular basis. Janse et al. , demonstrated circus movement
within the AV node as a basis for supraventricular tachycardia. They
employed multiple microelectrodes recording in the isolated rabbit heart
for their study.[13]
The animal model widely used for AV nodal re-entrant tachycardia is the
isolated rabbit heart preparation. However, this model does not mimic
the heart of patients suffering from AV nodal re-entrant tachycardia.[14] Wit et al., demonstrated
an in vitro model of paroxysmal supraventricular tachycardia.
They used in vitro preparation of rabbit heart atrium, including
the AV node and Bundle of His to evaluate the mechanism of paroxysmal
supraventricular tachycardia. Microelectrode recordings from the atrium
and AV node were observed. During sinus rhythm the atrial cycle was explored
with atrial premature depolarization.[15]
More et al., induced experimentally paroxysmal AV nodal tachycardia
in the dog.[16] Lin et al., experimentally
created atrioventricular node re-entrant tachycardia in the dog by surgery.
They blocked atrial impulses from the anterior input site to the AV node.[17]
Wu et al., used optical mapping in isolated canine atrioventricular
nodal re-entrant tachycardia.[18] This
study was performed to optically map Koch′s triangle and surrounding atrial tissue in an isolated canine AV nodal preparation. Multiple preferential AV nodal input pathways were observed in all preparation with continuous and discontinuous AV nodal function curves. AV nodal echo beats were induced in 54% (12/22) of preparations. The re-entrant circuit of the slow/fast echo beats (EB) (36%) started as a block in fast pathway and a delay in slow pathway (SP) conduction to the compact AV node, then excited from the AV node to the fast pathway and rapidly returned to the second pathway through the atrial tissue located at the base of Koch′s triangle. The re-entrant circuit of the fast/slow EB (9%, n=2) was in an opposite direction. In the slow/slow EB (9%,
n=2), anterograde conduction was over the intermediate pathway (IP) and
retrograde conduction was over the SP. Unidirectional conduction block
occurred at the junction between the AV node and its input pathways.
Conduction over the IP smoothed the transition from the FP to the SP,
resulting in a continuous AV nodal function curves. Complete or incomplete
echoes were induced in isolated preparations by this method.[19] Patterson et al., have
demonstrated that longitudinal dissociation within the posterior AV nodal
input can give rise to localized re-entry and AV nodal re-entrant tachycardia.[20]
Adenosine is a potent source to terminate AV nodal re-entrant tachycardia.
Hence, drugs such as adenosine can be screened by using these models.
Class Ic drugs can be screened for chronic use. AV nodal blockers [classes
II and IV (such as verapamil and diltiazem)] may be screened for acute
study.
Atrial flutter
Atrial flutter is a rapid regular atrial tachyarrhythmia that is less
common than the PSVTs or atrial fibrillation. It is observed only very
rarely in normal subjects but may occur at any age in the presence of
underlying abnormalities such as those secondary to mitral valve disease,
congenital heart disease, cardiomyopathies and less frequently coronary
artery disease.[21] Subgroups
at particularly high risk for developing atrial flutter are children,
adolescents and young adults, who have undergone corrective surgery for
complex congenital heart disease, most commonly transposition of the
great vessels, tetralogy of Fallot, or atrial septal defects.[22] Lewis et al., concluded
that atrial flutter was the result of circus movement in the atria.[23] Successful
animal preparations of atrial flutter have been developed over the years.
Some important animal models to induce atrial flutter are as follows.
Canine right atrial crush injury model
Gregory et al., used this method for induction of atrial flutter.
The atrial crush injury was made with a surgical clamp by lifting the
anterior portion of the right atrial plaque after recording baseline
sinus rhythm. The crush injury was placed on the right atrial free wall
parallel to and approximately 1.5 cm above the atrioventricular groove,
extending from the base of the right atrial appendage 1.5-2.5 cm posterior
towards the intercaval zone. The crush injury was typically 3-4 mm wide.
After right atrial crush injury, attempts were made to induce sustained
atrial flutter by programmed atrial stimulation introducing single (S1S2),
double (S1S2S3), or triple (S1S2S3S4)
premature beats to atrial refractoriness. Sustained atrial flutter was
defined as that lasting >10 min.[24]
Atrial flutter induced by acetylcholine (ACh) and rapid pacing in the dog
Wu et al., induce atrial flutter in the isolated blood perfused
canine heart. They produced episodes of rapid atrial flutter by continuous
infusion of ACh and rapid atrial pacing. They isolated canine right atria
and perfused it with 1-5 µM/l of ACh. Mapping of the endocardium
was done by using 477 bipolar electrodes with simultaneously recording
transmembrane potentials from the epicardium. The APD was measured during
regular pacing with cycle lengths of 300 ms. Atrial arrhythmia was induced
by a premature stimulus.[25]
Atrial flutter by aconitine
Scherf et al., [26] provoked
atrial flutter in anesthetized dogs by application of a few crystals
of aconitine or delphinine to the surface of the right atrium in the
appendix area near the head of the sinus node. Nwangw et al.,[27] used
aconitine (as an arrhythmogenic agent) to screen antiarrhythmic drugs
in mice. Dadkar and Bhattacharya recommended aconitine antagonism in
conscious mice as screening procedure.[28] Winslow
recommended it in anesthetized mice.[29] Also,
Winslow established the arrhythmogenic effects of aconitine in cats.[30]
Right atrial enlargement model of atrial flutter
Restivo et al., developed the canine model in which right atrial
enlargement was produced by banding of the pulmonary artery thereby producing
tricuspid regurgitation which may have a clinical counterpart in patients
with chronic obstructive pulmonary disease and tricuspid regurgitation.
It is now established that atrial flutter is due to a re-entrant wave
in the right atrium, and that a zone of slow conduction located inferiorly
and posterior in the right atrium is the target for catheter ablation.[31]
AV nodal blockade is a reliable mechanism to treat atrial flutter. Thus
adenosine like drugs, Ca2+ blockers
and beta-adrenergic blockers can be screened on chronic basis through
the models, which produce atrial flutter. Class Ia, Ib and class III
drugs can be screened for acute therapy.
Atrial fibrillation
The prevalence, presentation, clinical significance and long-term implications
of atrial fibrillation depend heavily upon the clinical circumstance
in which it occurs. Among the cross-sectional studies of prevalence,
there is a large gradient across age categories, ranging from less then
0.5% through the decades from 40 to 70 years and reaching rates in excess of 10% is
some beyond age 70.[32] The
haemodynamic consequences of atrial fibrillation are due to two factors:
(i) the loss of atrial systole may impair ventricular function in the
noncompliant ventricle (e.g. aortic stenosis, left ventricle hypertrophy
or the dilated ventricle with systolic dysfunction) and (ii) a rapid
ventricular rate encroaches upon diastolic filling of the left ventricle
and the coronary arteries.[33] The
risk of embolism and stroke is a long-term concern of special importance.
The left superior vena cava can be the arrhythmogenic source of AF. The
left superior vena cava (LSVC) is the embryological precursor of the
ligament of Marshall, which has been implicated in the initiation and
maintenance of atrial fibrillation. Rarely the LSVC may persist and has
been associated with some organized arrhythmias. Li. Fern reported five
patients in whom the LSVC was a source of ectopy, initiating atrial fibrillation.[34]
Atrial fibrillation by atrial ischaemia in dogs
Hani et al., induced AF by atrial ischaemia after occluding the
right intermediate atrial artery (a branch of the right coronary artery)
that perfuses the right atrial free wall. Atrial-arterial occlusion increased
the duration of AF induced by burst pacing from 57-32 to 803-214 sec
after 0.5 h of occlusion and to 887-209 sec after 3 h of occlusion. Prolonged
AF was induced in none of the 16 dogs under control nonischemic conditions,
7 of 16 dogs (44%, P<0.01) at 0.5-3 h after occlusion, and 5 of 13 dogs (38%, P<0.01)
3-5 h after occlusion.[35]
Pituitary adenylate cyclase activating polypeptide-27 (PACAP-27) induced biphasic chronotropic effect and atrial fibrillation
PACAP-27 causes negative chronotropic effect through postganglionic nerve
activation and it produces the positive chronotropic effect mediated
by PACAP receptors with an activation of nonadrenergic nonvasoactive
intestinal peptidergic nerves at least in part in the dog heart. Neurally
released acetylcholine induced by PACAP-27 participates in the induction
of atrial fibrillation.[36]
Atrial fibrillation in dogs by atrial burst pacing
Danshi et al., induced atrial fibrillation by atrial burst pacing
(10 Hz, 1 to 5 sec). Atrial fibrillation >20 min requiring electrical
cardioversion for termination was considered persistent. To estimate
mean atrial fibrillation duration, atrial fibrillation was induced 10
times if the duration was less than 10 minutes and 5 times if it was
10-20 min. If persistent atrial fibrillation was induced twice, no further
atrial fibrillation inductions were performed.[37]
Canine model of chronic atrial fibrillation
Thomas et al., has induced chronic atrial fibrillation in dogs
by creating moderate mitral regurgitation and rapidly pacing the right
atrium at 640 bpm for > 8 weeks. Chronic atrial fibrillation was established
with the combination of rapid atrial pacing and creation of moderate
regurgitation. Catheters were introduced into the left and right heart
of female mongrel dogs via femoral venous and arterial sheath. Baseline
hemodynamic measurements were recorded. A 7F steerable catheter with
a stiff 2 mm wire hook at its terminus was placed in the left ventricle
and manipulated until mitral chordae tendineae were ensnared and then
avulsed. An active fixation atrial J permanent pacemaker lead was placed
in the right atrial appendage by a jugular venous approach. The pacemakers
were programmed at the rate of 640 bpm or 400 bpm and an output of 2-3
times atrial diastolic threshold. After 6 weeks and weekly thereafter,
the pacemakers were reprogrammed to low rates and to subthreshold outputs.
After 24 h without pacing, a six lead surface ECG was obtained to verify
the presence of atrial fibrillation.[38]
Vagal atrial fibrillation
Parasympathetic stimulation has been used for decades for the induction
and maintenance of atrial fibrillation in experimental protocols.[39] Parasympathetic
stimulation dramatically shortens the atrial effective refractory period
thereby decreasing the wavelength of atrial excitation wave fronts. The
shorter the wavelength, the higher is the probability that multiple re-entrant
circuits can exist simultaneously in the atrial myocardium; the presence
of these multiple circuits, in turn, increases the stability of atrial
fibrillation.[40]
For cervical vagal nerve stimulation, the cervical vagosympathetic trunks
were cut, and stainless steel wires were introduced in the cranial end
of the vagosympathetic trunk. Stimulation of both the vagi was performed
with separate isolated constant current sources (SS-202J, Nihon Kohden)
driven by a programmable stimulator (SEN-7203, Nihon Kohden). During
bilateral vagal stimulation, AF was induced by atrial extrastimulation
using a digital programmable stimulator (SEC-2102, Nihon Kohden). AF
lasting more than 10 min was repeatedly induced in dog.[41]
Atrial fibrillation in the isolated Langendorff-perfused rabbit heart
Atrial arrhythmias frequently occur under conditions associated with
atrial dilation.[42],[43] In
patients with acute myocardial infarction, the onset of atrial arrhythmias
is thought to be related to the elevated left ventricular end diastolic
pressure, resulting in stretch of the atrial wall.[42] In
the Langendorff perfused rabbit heart, the interatrial septum was perforated,
and after occlusion of the caval and pulmonary veins, bilateral pressure
was increased by raising the level of an outflow cannula in the pulmonary
artery. Right and left arterial effective refractory periods, monophasic
action potentials and inducibility of atrial flutter by single premature
stimuli were measured as a function of atrial pressure. Increasing the
atrial pressure from 0.5±0.7 to 16.2±2.2 cm H2O
resulted in a progressive shortening of the right atrial effective refractory
period (AERP) from 82.2±9.8 to 48.0±5.1 ms. In the left atrium, an increase in pressure up to 7.4±0.3
cm H2O had no effect on the AERP. At higher pressures, however,
the left AERP also shortened, from 67.5±7.5 to 49.3±2.0 ms. The duration of monophasic action potentials (MAP) also decreased by an increase in atrial pressure, showing a high correlation with the shortening in AERP (r=0.94, P<0.01).
All these changes were completely reversible within 3 min after release
of the atrial stretch. Dilatation of the atria was a major determinant
for the vulnerability to AF.[43]
Atrial fibrillation by fibrillation pacemaker
Goats were chronically instrumented with multiple electrodes sutured
to the epicardium of both atria. Two to three weeks after implantation,
the animals were connected to a fibrillation pacemaker which artificially
maintained atrial fibrillation. Control episodes of AF were short lasting
(6±3 sec) but artificial maintenance of AF resulted in a progressive increase in the duration of AF to become sustained (>24 h) after 7.1±4.8
days.[44]
Atrial fibrillation by rapid atrial pacing and acetylcholine
Burashnikov et al., induced atrial fibrillation in isolated sheep
hearts by burst rapid pacing from the epicardial surface of either the
right atria after the addition of acetylcholine (1 µM/L) to the
perfusate. Transmembrane action potentials, pseudo-ECG (pseudo-ECGs are
constructed from optical recordings by integrating the transmembrane
fluorescence signal over the left and right halves of the mapped region
and taking the difference) and tension development were recorded.[45] Allan et al., recorded
the optical recordings of atrial movements to demonstrate wave propagation
and lines of block, which changed on a beat to beat basis.[46]
Atrial fibrillation by aconitine
The plant alkaloid aconitine persistently activates sodium channels.
Nakayama et al., compared the effects of various beta adrenergic
blocking agents with known antiarrhythmics on aconitine arrhythmia.[47] They
produced supraventricular arrhythmias by topical application of aconitine
in a small cup placed on the right atrium of dogs. Yamamoto et al., used
urethane-anesthetized rats under artificial respiration with tubocurarine
pretreatment. After thoracotomy and incision of the pericardium, a piece
of filter paper soaked with aconitine solution was applied to the right
atrium. Test drugs were applied by continuous i.v. infusion. ECG lead
II and intra atrial ECG were monitored.[48]
Therapy of atrial fibrillation is almost same as atrial flutter. Thus
choice of drugs, which can be screened through these models of atrial
fibrillation, is same as atrial flutter.
Ventricular fibrillation
Porcine model of VF
Iyad et al., used this model to show ventricular anti-fibrillation
activity of cariporide (sodium-hydrogen exchanger isoform-1 inhibitor).
A 5F pacing electrode was advanced through the right cephalic vein into
the right ventricle for induction of VF. VF was induced by an alternating
current (1-5 mA) delivered to the right ventricular endocardium, and
mechanical ventilation was discontinued. The effects on ischemic contracture
were investigated by the use of transesophageal echocardiography (TEE).
The effects on APD and ventricular ectopic activity were investigated
by the use of a monophasic action potential (MAP) recording/pacing catheter.[49]
Ventricular fibrillation induction by 60-Hz alternating current in isolated swine right ventricle.[50]
Voroshilovsky et al., studied seven isolated perfused swine right
ventricle in vitro .
The action potential duration restitution curve was determined. Alternating
current captured the right ventricles at 100± 65 µA, which is significantly lower than the direct current pacing threshold (0.77 ± 0.45 mA P<0.05). Alternating current induced ventricular tachycardia or ventricular fibrillation at 477 ± 266 µA, when the stimulated response to alternating current had (1) short activation cycle lengths (128±14 ms), (2) short diastolic intervals (16±9
ms) and (3) short diastolic intervals associated with a steep action
potential duration restitution curve. Optical mapping studies showed
that during rapid ventricular stimulation by alternating current, a wave
front might encounter the refractory tail of an earlier wave front, resulting
in the formation of a wave break and ventricular fibrillation.
Ventricular arrhythmia by various chemicals
Vogel and Vogel described a method in which they administered continuous
aconitine infusion into the saphenous vein to produce ventricular arrhythmias.[51] Vaille et al., used
CaCl2 continuous i.v. infusion screening method in rats for
evaluation of antiarrhythmic calcium antagonists.[52] Lawson
induced ventricular fibrillation in mice by using chloroform as an antiarrhythmic
agent.[53] Tripathi and
Thomas produced ventricular tachycardia in rats and guinea pigs by exposing
the animals to benzene vapours for 2 min followed by an intravenous adrenaline
injection.[54] Digoxin a
cardiac glycoside, if given in overdose produces ventricular extrasystoles,
ventricular fibrillation, and finally death. Nagasawa et al., used
digitalis induced arrhythmia model in dogs for screening of a new antiarrhythmic
drug which was Na+/Ca+[2] exchange
inhibitor.[55] Sono et al.
induced ventricular fibrillation by isoprenaline in isolated rat hearts.[56] Tomokazu et al., used
ouabain or strophanthin K, which is a cardiac glycoside as an arrhythmogenic
substance.[57] Ouabain induced
ventricular tachycardia and multifocal ventricular arrhythmias in dogs.[58] Ra et al., demonstrated
a modified method for the production of cardiac arrhythmias by ouabain
in anesthetized cats.[59] Takei
described experimental arrhythmia in guinea pigs induced by grayanotoxin-I,
a biologically active diterpenoid from the plant family of Ericaceae.[60]
Ischaemia-induced ventricular arrhythmia
Cardiac ischaemia leading to myocardial infarction is a most common cause
of morbidity and mortality. Chemical or surgical interventions allow
the recovery of the ischaemic myocardium by restoration of blood flow
or reperfusion. This reperfusion, however, is known to be associated
with ventricular arrhythmias and myocardial dysfunction that can lead
to severe cardiac impairment and cell death.[61], [62] Reactive
oxygen species (ROS) such as superoxide anion, hydrogen peroxide, hydroxyl
radical, and singlet oxygen have been implicated as important factors
in the pathogenesis of cellular injury in the postischemic heart. [63],[64],[65]
Coronary artery occlusion and reperfusion in the isolated perfused rat
heart, was widely used as a model for assessment of antifibrillatory
action of antiarrhythmic agents.[66] Later,
coronary occlusion/reperfusion arrhythmias have been shown in anesthetized
animals. Clark et al., demonstrated coronary artery ligation
in anesthetized rats as a method for the production of experimental dysrhythmias
and for the determination of infract size.[67] Harris et al., studied
influences of hypothermia, cold and isolation stress on the severity
of coronary artery ligation-induced arrhythmias in rats.[68] Bernier et al., used
reperfusion-induced arrhythmias and studied oxygen derived free radicals,
which causes myocardial infarction and ventricular arrhythmia, in isolated
perfused rat heart.[69] Lepran et al., used
the coronary artery ligation technique in rats after 7-10 days of surgery.
They placed a loose silk loop around the left coronary artery and passed
the threshold through a cylinder-shaped polyethylene tube outside the
thorax. The loose ligature was tightened and arrhythmia record by ECG.[70] Lubbe et al., used
coronary artery occlusion and reperfusion in isolated perfused rat heart
for assessment of antifibrillatory action of antiarrhythmic agents.[71] Macleod et al., induced
arrhythmia by ischaemia and reperfusion in conscious and anesthetized
rats, and they studied the effect on epicardial intracellular action
potentials.[72]
In the ischaemia-reperfusion model different parameters can be evaluated,
such as mortality, haemodynamic parameter, ventricular extrasystoles,
ventricular tachycardia, ventricular fibrillation and infract size. [73],[74],[75] The
number of ventricular premature beats, ventricular tachycardia and ventricular
fibrillation are counted in the occlusion and reperfusion periods and
evaluated according to the guidelines of Lambeth convention.[76] Capasso et al., reported
heterogeneity of ventricular remodelling after acute myocardial infarction
in rats which was produced by the ischaemia-reperfusion technique.[77] Ruben et al., studied
the distribution of extracellular potassium and its relation to electrophysiological
changes during acute myocardial ischaemia in the isolated perfused porcine
heart.[78] Bradykinin perfusion
reduced the incidence of ventricular fibrillation and reduced the release
of cytosolic enzyme and preserved glycogen stores.[79] Ventricular
arrhythmias occurring secondary to impeded myocardial perfusion is the
cause of death in more then one-half of the subjects associated with
myocardial infarction and is also an important cause of sudden cardiac
death.[80] Two distinct
phases of ventricular arrhythmias occur during the first 30 minutes after
induction of regional ischaemia by acute occlusion of a coronary artery
in canine and porcine heart.[81],[82] In
the first phase, Ia (up to approximately 8-10 min of coronary occlusion),
there is a rapid change in electrical membrane properties associated
with metabolic acidification (anaerobic glycolysis), and cellular loss
and extracellular accumulation of [K+].[83] The
impacts of these changes are a rapid depolarization of the ischemic myocytes,
and a loss of amplitude and duration of the transmembrane potential.
In second phase, Ib, which occurs after 10-15 min of occlusion is related
to the electrical uncoupling of the myocytes resulting in smaller size
of circus movement.[84] William et al., reported
that the Ib phase of ventricular arrhythmias in ischemic in situ porcine
heart is related to changes in cell-to-cell electrical coupling.[85] Borrett et al., described
a myocardial ischaemia model for arrhythmia in rabbits.[86] Vara et al., induced
ventricular fibrillation by coronary occlusion during hypothermia in
dogs.[87] Baboon open chest
model of myocardial ischaemia was described by Premaratne et al., in
which a 2 h ischaemia period is followed by 22 h of reperfusion.[88] Naslund et al., presented
a closed chest model in pigs. In this model, occlusion was induced in
closed chest, pentobarbitone anaesthetized, and mechanically ventilated
pigs by injection of a 2 mm ball into a preselected coronary artery.
Reperfusion was achieved by retraction of the ball via an attached filament.
Ventricular arrhythmia during exercise by ischaemia
Ventricular fibrillation due to myocardial ischaemia during exercise
is the model which resemble most closely the situation in coronary patients.[90],[91] In
this model a major surgery is done in dogs, in which transducers are
fixed in body followed by two-stage LAD ligation. After 28 days, animals
were prepared for test, in which animals are allowed to walk on a motor
driven treadmill. The animals run on the treadmill simultaneously the
workload was increased in every 3 min for 18 min.[92]
Stretch-induced arrhythmias in isolated canine ventricle
It is commonly accepted that serious ventricular arrhythmias are caused
by abnormalities in impulse formation and conduction.[1] These
electrophysiological mechanism fail to explain why lethal arrhythmia
most commonly arise in patients with severe heart failure and dilated
ventricles.[93],[94] David et al., presented
a hypothesis that alternation in loading conditions and muscle length
influenced the electrophysiology of ventricular myocardium and these
alterations might play a role in arrhythmogenesis in globally dilated
or dyskinetic ventricles. To test the hypothesis that stretch can initiate
arrhythmias in normal myocardium, the response to graded mechanical stretch
was studied in seven isolated blood perfused canine ventricles. After
eight conditioning contraction produced by His bundle pacing (2 Hz),
global stretch of the ventricle was produced by a servo-controlled pump
that abruptly increased ventricle volume by a precise amount during early
diastole and then returned ventricular volume to the initial holding
volume. The probability of a stretch-induced arrhythmia was determined
from multiple alternating sequences in which a stretch of known amplitude
or no stretch was delivered.[95],[96]
Model for sudden cardiac death
Male mongrel dogs weighing 14-22 kg are used for this model. Programmed
electrical stimulation is performed between days 3 and 5 after the induction
of anterior myocardial infarction by occlusion/perfusion on the left
anterior descending coronary artery. A direct anodal 15 µA current
from a 9V nickel-cadmium battery was passed through a 250 Ohm resister
and applied to the electrode in the lumen of the left circumflex coronary
artery. After 24 h of constant anodal current or development of ventricular
fibrillation, the animals were killed, the hearts were excised and the
thrombus mass removed and weighed.[97],[98]
Canine model of two-stage ligation
Harris showed that mortality in dogs after coronary occlusion with a
two-stage ligation procedure was lower than with one-stage ligation.
Left descending coronary artery is partially occluded for 30 min, after
which total ligation is performed. This model resembles late arrhythmias
occurring in post-infarction patients.[99] Akira et al., used
this model to check antiarrhythmic activity of a new compound.[100] Dubray et al .
presented methods for producing experimental complete atrioventricular
block in dogs.[101]
Drugs, which can be screened for ventricular tachycardia, are class Ia,
Ib and class III drugs for both acute and chronic therapy. Ventricular
fibrillation is a result of disorganized re-entry and classes Ia and
Ib drugs are useful for acute therapy and can be screened for acute uses.
For chronic use classes I-III can be screened.
Transgenic mice for arrhythmia Genetically engineered animal models hold promise for understanding the pathophysiology of mutations that cause human disease.[102],[103] In the last decade various artificial mutation in mice genotype yielded a number of transgenic mice, which are useful in screening for anti-arrhythmics. Berul et al., used cardiac electrophysiology method to study mice harbouring an α-myosin heavy chain Arg 43 Gln missense mutation (α-MHC 403/+), which resulted in histological and haemodynamic abnormalities characteristic of familial hypertrophy and sudden death of uncertain etiology during exercise.[104] Wu. et al., studied a mouse model of cardiac hypertrophy attributable to transgenic over-expression of a constitutively active form of CaMK IV that also has increased endogenous CaMK II activity. ECG telemetered transgenic mice had significantly more arrhythmias then wild type littermate controls at baseline.[105] The KCNE 1 gene encodes a channel regulator IsK which, in association with the KvLQT1K+ channel protein, determines the slow component of the delayed rectifier current. Charpentier et al., investigated the cellular electrophysiological characteristics of adult KCNE1 knockout mouse hearts by means of the standard microelectrode technique. They concluded that invalidation of the mouse KCNE1 gene by homologous recombination leads to a mild cardiac phenotype at the cellular level. Berul et al., presented a mouse model of dilated cardiomyopathy resulting from a homozygous mutation in the myosin-binding protein C (MyBY- Ct/t). They also presented a model of familial hypertrophic cardiomyopathy due to heterozygous mutation in the same gene (MyBP-C) that were used to characterize the electrophysiological phenotype and correlate ′vulnerability to arrhythmia′ with quantitative histopathological changes.[107] As described earlier mutations in the γ2 subunit (PRKAG2) of AMP activated protein kinase produce an unusual human cardiomyopathy characterized by ventricular hypertrophy and electrophysiological abnormalities such as: Wolf-Parkinson White syndrome and programmed degenerative conduction system disease. Mutations of the K+ channel genes HERG and KVLQT 1 cause the autosomal dominant long QT syndrome, presumably by interfering with the cardiac currents Ikr and Iks. [108],[109],[110] The precise mechanism by which the mutations lead to QT prolongation and arrhythmias is uncertain. An N-terminal fragment including the first trans-membrane segment of the rat delayed rectifier K+ channel Kv 1.1 (Kv1.1 N 206 Tag), co-assembles with other K+ channels of the Kv1 subfamily in vitro , inhibits the currents encoded by Kv 1.5 in a dominant negative manner when co-expressed in Xenopus oocytes, and traps Kv 1.5 polypeptide in the endoplasmic reticulum of GH3 cells.[111] Barry et al., reported that transgenic mice over-expressing Kv 1.1 n 26 Tag in the heart have a prolonged QT interval and ventricular tachycardia.[112] Nkx 2.5 is a conserved homeodomain containing transcription factor essential for early cardiac development. Both prenatal and postnatal over-expression of DNA of nonbinding mutant Nkx 2.5 are associated with AV conduction malfunction and heart failure; however, more profound progressive electrophysiologic defects are seen when this mutation expresses during fetal and neonatal periods. These conduction abnormalities may contribute to the lethal heart failure and early mortality evident in DNA nonbinding mutant Nkx 2.5 mice.[113] Lande et al., evaluated a transgenic mouse over-expressing a dominant negative KvLQT 1 isoform, as an in vivo screening model for IKr blocking drugs. They concluded that KvLQT invalidated transgenic mice discriminates in vivo drugs that blocks IKr from drugs that block the transient outward current, the sodium current or the calcium current.[114] Transgenic mice over-expressing the inflammatory cytokine tumour necrosis factor TNF-α (TNF-α mice) in the heart develop a progressive heart failure syndrome characterized by biventricular dilation, decreased ejection fraction, atrial and ventricular arrhythmias on ambulatory telemetry monitoring, and decreased survival compared with nontransgenic litter mates. These transgenic animals are more prone to re-entrant arrhythmia.[115]
Antiarrhythmic drugs and screening models Antiarrhythmic drugs generally affect arrhythmia by modulating conduction velocity, or effective refractory period or both. Conduction velocity, depends on the passive electrical properties of cardiac tissue, also is a characteristic of the Na+ channels and Ca+ 2sub channels. Antiarrhythmic drugs that prolongs the action potential duration, and thereby the refractory period are effective against re-entry arrhythmias in two ways: by prolonging the wavelength [the product of (a) refractory period and (b) conduction velocity.[116] The initiation of a re-entrant arrhythmia by a premature impulse may be prevented[117] or an existing arrhythmia may terminate because the wavelength becomes too large with respect to the re-entrant circuit, so that by closing the excitable gap, the head of the re-entrant wavelength will hit the wall of refractoriness and propagation steps.[118] It is well known that there is significant difference in effective refractory period of different species. [119],[120],[121],[122] Depending on action potential duration and effective refractory period selection of animal could be one important aspect. Porcine ventricle myocardium appears to have a diastolic interval similar to that in human ventricle.[119] In contrast to all other species, it may be appreciated that in the rat there is no shortening of refractory periods at the shorter cycle length and rat ventricle is not the first choice if one aims at filling up the diastolic interval by means of a class I or class III antiarrhythmic agent.[120],[121] The rabbit is often used for electrophysiological research, probably because it constitutes a reasonable compromise in terms of cardiac dimensions, basic electrophysiological characteristics and cost.[122],[123] There are clear species differences that determine arrhythmogenesis. These differences should also be considered while choosing an animal model for arrhythmia corresponding to antiarrhythmic agent.
Conclusion
Although no animal model can accurately resemble with human disease condition
and species differences also exist, close similarities with humans suffering
from or threatened by arrhythmias can be developed by selecting appropriate
model and species. Rather than a single model or experimental technique,
combinations of investigations, like isolated heart (Langendorff arrangement
or working heart), whole hearts in anesthetized or conscious animals, excised
cardiac preparations, testing the function of molecules involved in electrical
excitation, single cardiac cell preparation, can be performed.
References
| 1. | Keating MT, Sanguinetti MC. Molecular and cellular mechanisms of cardiac arrhythmias. Cell 2001;104:569-80. Back to cited text no. 1 [PUBMED] [FULLTEXT] |
| 2. | Vaughan- Williams EM. Classification of antiarrhythmic drugs. In: SandÖe E, Flensted-Jensen E, Olesen KH, editors. Symposium on cardiac arrthmias. Elsinore , Denmark: AB Astra, S φdertδlje, Sweden. 1970. Back to cited text no. 2 |
| 3. | Keating L, Morris FP, Brady WJ. Electrocardiographic features of Wolff-Parkinson-White syndrome. Emerg Med J 2003;20:491-3. Back to cited text no. 3 [PUBMED] [FULLTEXT] |
| 4. | Wellus HJJ. The electrophysiological properties of the accessory pathways in the Wolf-Parkinson-White syndrome. In: Welleus H, Lie KI, Jause MJ, editors. The conduction system of the heart. Leiden: Stenfert Kroese;1988. Back to cited text no. 4 |
| 5. | Boineau JP, Moore EN. Evidence for propagation of activation across an accessory atrioventricular connection in type A and type B preexcitetion. Circulation 1970;41:375-97. Back to cited text no. 5 |
| 6. | Kemp BE, Mitchelhill Kl, Staplenton D. Dealing with energy demand: the AMP activated protein kinase. Trends Biochem Sci 1999:24:22-25 Back to cited text no. 6 |
| 7. | Gollob MH, Green Ms, Tang AS. Identification of a gene responsible for familial Wolf-Parkinson-White syndrome. N Eng J Med 2001;344:1823-31. Back to cited text no. 7 |
| 8. | Arad M, Moskowitz IP, Patel VV, Ahmad F. Transgenic mice overexpressing mutant PRKAG2 define the cause of Wolf-Parkinson-White syndrome in glycogen storage cardiomyopathy. Circulation 2003;107:2850-2856. Back to cited text no. 8 |
| 9. | Efimov IR, Nikolski VP, Rothenberg F, Greener ID, Li J, Dobrzynski H, et al . Structure-function relationship in the AV junction. The Anatomical Record 2004;280:952-65. Back to cited text no. 9 |
| 10. | Bharati S. Anatomic-morphologic relations between AV nodal structure and function in the normal and diseased heart. In: Mazgalev TN, Tchou PJ, editors. Atrial-AV nodal electrophysiology: A view from the millenium. Armonk: Futura; 2000. 25-48. Back to cited text no. 10 |
| 11. | Cox JL, Holman HL, Cain ME. Cryosurgical treatmrnt of atrioventricular node reentrant tachycardia. Circulation 1987;76:1329-76. Back to cited text no. 11 |
| 12. | Sung RJ, Waxman HL, Saksen S, Juma Z. Sequence of retrograde atrial activation in patients with dual atrioventricular nodal pathways. Circulation 1981;64:1059-67. Back to cited text no. 12 |
| 13. | Janse MJ, Van Capelle FJL, Freud GE, Durrer D. Circus movement within the A-V node as a basis for supraventricular tachycardia as shown by multiple microelectrode recording in the isolated rabbit heart. Circ Res 1971;28:403-13. Back to cited text no. 13 |
| 14. | Mazgalev TN, Ho SY, Anderson RH. Anatomic-electrophysiological correlations concerning the pathways for atrioventricular conduction. Circulation 2001;103:2660-7. Back to cited text no. 14 [PUBMED] [FULLTEXT] |
| 15. | Wit AL, Goldreyer BN, Damato AN. AN in vitro model of paroxysmal supraventricular tachycardia. Circulation 1971;43:862-75. Back to cited text no. 15 [PUBMED] |
| 16. | More GK,Cohen W. Vick RL. Experimentally induced paroxysmal A-V nodal tachycardia in the dog. A 'case report'. Am Heart J 1963;65:87-92. Back to cited text no. 16 |
| 17. | Lin FY, Lo HM, Cheng JJ. Experimentally created atrioventricular node reentrant tachycardia in the dog: Evidence of a brake system for nodal reentry in the anterior interatrial septum. J Am Coll Cardiol 1993;22:1541-7. Back to cited text no. 17 [PUBMED] |
| 18. | Wu J, Wu J, Olgin J, Miller JM, Douglas P. Zips; Mechanism underlying the reentrant circuit of atrioventricular nodal reentrant tachycardia in isolated canine atrioventricular nodal preparation using optical mapping. Circ Res 2001;88:1189. Back to cited text no. 18 |
| 19. | Loh P, Siew Yen ho, Kawara T, Hauer RNW, Janse MJ. Reentrant circuits in the canine atrioventricular node during atrial and ventricular echoes. Circulation 2003;108:231. Back to cited text no. 19 |
| 20. | Patterson E, Scherlog BJ. Longitudinal dissociation within the posterior AV nodal input of the rabbit. A substrate for AV nodal reentry. Circulation 1999;99:143-55. Back to cited text no. 20 |
| 21. | Lee K, Yang Y, Scheinman M. Atrial flutter: A review of its history, mechanisms, clinical features, and current therapy. Current Problems in Cardiology 2003;30:121-67. Back to cited text no. 21 |
| 22. | Daoud EG, Fred Morady MD. Pathophysiology of atrial flutter. Ann Rev Med 1998;49:77-83. Back to cited text no. 22 |
| 23. | Ewis T, Drury AN, I Iiescu CC. A demonstration of circus movement in clinical flutter of the auricles. Heart 1921;8:341-57. Back to cited text no. 23 |
| 24. | Feld GK, Rad FS. Mechanism of double potentials recorded during sustained atrial flutter in the canine right atrial crush injury model. Circulation 1992;86:628-41. Back to cited text no. 24 |
| 25. | Wu TJ, Kim YH, Yashima M, Athill CA, Ting CT, Karagueuzian HS, et al . Progressive action potential duration shortening and the conversion from atrial flutter to atrial fibrillation in the isolated canine right atrium. J Am Coll Cardiol 2001;38:1757-65. Back to cited text no. 25 |
| 26. | Scherf D, Blumenteld S, Taner D, Yildiz M. The effect of diphenyldantoin on atrial flutter and fibrillation provoked by focal application of aconitine or delphine. Am Heart J 1960;60:936-47. Back to cited text no. 26 |
| 27. | Nwangwa PV, Holcslaw TL, Stohs JS. A rapid in vivo technique for preliminary screning of antiarrhythmic agents in mice. Arch Int Pharmacodyn 1977;229: 219-26. Back to cited text no. 27 |
| 28. | Dadkar NK, Bhattacharya BK. A rapid screening procedure for antiarrhythmic activity in the mouse. Arch Int Pharmacodyn Ther 1974;212:297-301. Back to cited text no. 28 [PUBMED] |
| 29. | Winslow E. Evaluation of antagonism of aconitine induced dysrhythmia in mice as a method of decting and assassing antidysrhythmic activity. Br J Pharmac 1980;71:615-22. Back to cited text no. 29 [PUBMED] |
| 30. | Winslow E. Hemodynamic and arrhythmogenic effects of aconitine applied to the left atria of anesthetized cats. Effects of amiodarone and atropine. J Cardiovas Pharmac 1981;3:87-100. Back to cited text no. 30 |
| 31. | Restivo M, Hegazy M, El-Hamami M, Yin H, Caref EB, Assadi MA, et al . Efficacy of azimilide and dofetilide in the dog right atrial enlargement model of atrial flutter. J Cardiovasc Electrophysiol 2001;12:1018-24. Back to cited text no. 31 |
| 32. | Cairns JA, Connolly ST. Nonrheumatic atrial fibrillation: Risk of stroke and role of antithrombotic therapy. Circulation 1991;84:469-81. Back to cited text no. 32 |
| 33. | Myerburg RJ, Kessler KM, Castellanes A. Recognition, clinical assessment and management of arrhythmias and conduction disturbences. Hurst's the Heart. 8th ed. McGraw Hill, Inc. Health Profession Division 1994. Back to cited text no. 33 |
| 34. | Hsu LF, Jais P, Keane D, Wharton JM, Deisenhofer I. Atrial fibrillation originating from persistent left supeior vena cava. Circulation 2004;109:828-32. Back to cited text no. 34 |
| 35. | Sinno H, Derakhchan K, Libersan D, Merhi Y, Leung TK, Nattle S. Atrial ischaemia promotes atrial fibrillation in dogs. Circulation 2003;107:1930-6. Back to cited text no. 35 |
| 36. | Hirose M, Furukawa Y, Nagashima Y, Lakhe M, Chiba S. Pitutary adenylate cyclase activating polypeptide-27 causes a biphasic chronotropic effect and atrial fibrillation in autonomically decentralized anesthetized dogs. J Pharmacol Exp Ther 1997;282:278-87. Back to cited text no. 36 |
| 37. | Li D, Shinagawa K, Pang L, Leung TK, Cardin S, Wang Z, Nattel S. Effects of angiotensin-converting enzyme inhibition on the development of the atrial fibrillation substrate in dogs with ventricular tachypacing-induced congestive heart failure. Circulation 2001,104:2608-14. Back to cited text no. 37 |
| 38. | Everett TH 4th, Li H, Mangrum JM, McRury ID, Mitchell MA, Redick JA, et al. Electrical, morphological, and ultrastructural remodeling and reverse remodeling in a canine model of chronic atrial fibrillation. Circulation 2000;102:1454-60. Back to cited text no. 38 |
| 39. | Allessie MA, Lammers WJEP, Smeets JRLM. Experimental evaluation of Moc's multiple wavelet hypothesis of atrial fibrillation. In: Zipes DP, Jalife J, editors. Cardiac Arrhythmias. New York: Grune and Stratton;1985. Back to cited text no. 39 |
| 40. | Schauerte P, Scherlag BJ, Jan P, Scherlag MA. Catheter ablation of cardiac anatomic nerves for prevention of vagal atrial fibrillation. Circulation 2000;102:2774-80. Back to cited text no. 40 |
| 41. | Nagasawa H, Fujiki A, Fujikura N, Matsuda T, Yamashita T, Inoue H. Effects of a novel class III antiarrhythmic agent, NIP-142, on canine atrial fibrillation and flutter. Circ J 2002;66 : 185-91. Back to cited text no. 41 |
| 42. | Manyari DE, Patterson C, Johnson D, Melendez L, Cape RDT. Atrial and ventricular arrhythmias in asymptometic active elderly subjects: correlaion with left atrial size and left ventricular mass. AM Heart J 1990;119:1069-76. Back to cited text no. 42 |
| 43. | Ravelli F, Allessie M. Effect of atrial dilation on refractory period and vulnerability to atrial fibrillstion in the isolated langendroff perfused rabbit heart. Circulation 1997;96:1686-95. Back to cited text no. 43 [PUBMED] [FULLTEXT] |
| 44. | Wijffels MC, Kirchhot CJ, Dorland R, Allessie MA. Arial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation 1995;92:1954-68. Back to cited text no. 44 |
| 45. | Burashnikov A, Antzelevitch C. Reinduction of atrial fibrillation immediately after termination of the arrhythmia is mediated by late phase early afterdepolarization-induced triggered activity. Circulation 2003;107:2355-60. Back to cited text no. 45 [PUBMED] [FULLTEXT] |
| 46. | Sknes AC, Mandapati R, Berenfeld O, Davidenko JM, Jalife J. Spatiotemporal periodicity during atrial fibrillation in the isolated sheep heart. Circulation 1998;98:1236-48. Back to cited text no. 46 |
| 47. | Nakayama K, Oshima T, Kumakura S, Hashimoto K. Comparison of the effects of various β-adrenergic blocking agents with known antiarrhythmic drugs on aconitine-arrhythmia produced by the cup method. Eur J Pharmacol 1971;14:9-18. Back to cited text no. 47 [PUBMED] |
| 48. | Yamamoto T, Hosoki K, Karasawa T. Anti-arrhythmic effects of a new calcium antagonist, Monopetil, AJ-2615, in experimental arrhythmic models. Clin Exper Pharmacol Physiol 1993;20:497-500. Back to cited text no. 48 |
| 49. | Ayoub IM, Kolsrova J, Yi Z, Trevedi A, Desmukh H, Lubell DL, et al . Sodium hydrogen exchange inhibition during ventricular fibrillation: Beneficial effects on ischemic contracture, action potential duration, reperfusion arrhythmias, myocardial function and resuscitability. Circulation 2003;107:1804-9. Back to cited text no. 49 |
| 50. | Voroshilovskey O, Qu Z, Lee MH, Ohara T, Fishbein GA, Lun HA, et al . Mechanisms of ventricular fibrillation induction by 60 hz alternating current in isolated swine right ventricle. Circulation 2000;102:1569-74. Back to cited text no. 50 |
| 51. | Vogel GH, Vogel WH, Scholkens BA, Sandow J, Muller G, Vogel WF, editors. Drug discovery and evaluation. Pharmacological assays. Berlin: Springer-Verlag; 2nd ed. 2002. Back to cited text no. 51 |
| 52. | Vaille A, Scotto Di Tella AM, Maldonado J, Vanelle P, Selectivity of a CaCl2 continuous infusion screening method in rats. Methods Find Exp Clin Pharmacol 1992;14:183-7. Back to cited text no. 52 |
| 53. | Lawson JW. Antiarrhythmic activity of some isoquiniline derivetives determined by a rapid screening procedure in the mouse. J Pharmacol Exp Ther 1968;160:22-31. Back to cited text no. 53 |
| 54. | Tripathi RM, Thomas GP. A simple method for production of ventricular tachycardia in the rat and guinea pig. J Pharmacol Methods 1986;15:279-82. Back to cited text no. 54 |
| 55. | Nagasawa Y, Zhu BM, Chen J, Kamiya K, Miyamoto S, Hashimoto K . Effects of SEA0400, a Na+/Ca2+ exchange inhibitor, on ventricular arrhythmias in the in vivo dogs. Eur J Pharmacol 2005;506:249-55. Back to cited text no. 55 |
| 56. | Sono K, Akimoto Y, Magaribuchi T, Kurahashi K, Hujiwara A. A new model of ventricular fibrillation induced by isoprenaline and catechol-o-methyl transferase inhibitor at high perfusion temperature in the isolated rat heart. J Pharmacol Methods 1985;14:249-54. Back to cited text no. 56 |
| 57. | Watano T, Harada Y, Harada K, Noriyasu N. Effect of Na+/Ca2+ exchange inhibitor, KB-R7943 on ouabain-induced arrhythmias in guinea-pigs. Br J Pharmacol 1999;127:1846-50. Back to cited text no. 57 |
| 58. | Duce BR, Garerg L, Johansson B. The effect of propranolol and the dextro and leavo isomers of H 56/28 upon ouabain induced ventricular tachycardia in anesthetized dogs. Acta Pharmacol Toxicol 1967;25:41-9. Back to cited text no. 58 |
| 59. | Rao TS, Seth SD, Nayar U, Manchanda SC. Modified method for the production of cardiac arrhythmias by ouabain in anesthetized cats. J Pharmacol Methods 1988;20:255-63. Back to cited text no. 59 |
| 60. | Takei. Grayanotoxin-I induced experimental arrhythmia in guinea pig. J Aichi Med Univ Assoc 1994;22:495-512. Back to cited text no. 60 |
| 61. | Moens AL, Claeys MJ, Timmermans JP, Vrints CJ. Myocardial ischaemia/reperfusion injury, a clinical view on a complex pathophysiological process. Int J Cardiol 2005;100:179-90. Back to cited text no. 61 |
| 62. | Gross GJ, Kersten JR, Warltie DC. Mechanisms of postischemic contractile dysfunction. Ann Thorac Sur 1999;68:1898-904. Back to cited text no. 62 |
| 63. | Webb A, Bond R, McLean P, Uppal R, Benjamin N, Ahluwalia A. Reduction of nitrite to nitric oxide during ischaemia protects against myocardial ischaemia-reperfusion damage. PNAS 2004;101:13683-8. Back to cited text no. 63 |
| 64. | Bolli R, Marban E. Molecular and cellular mechanisms of myocardial stunning. Physiol Rev 1999;79:609-34. Back to cited text no. 64 |
| 65. | Homans DC, Asinger R, Pavek T, Crampton M, Lindstrom P, Peterson D, et al . Effect of superoxide dismutase and catalase on regional dysfunction after exercise-induced ischaemia. Am J Physiol 1992;263:392-8. Back to cited text no. 65 |
| 66. | Lubbe WF, Daries PS, Opie LH. Ventricular arrhythmias associated with coronary artery occlusion and reperfusion in the isolated perfused rat heart: A model for assessment of antifibrillatory action of antiarrhythmic agents. Cardiovasc Res 1978;12:212-20. Back to cited text no. 66 |
| 67. | Clark C, Forman MI, Kane KA, Mc Donald FM, Parratt JR. Coronary artery ligation in anesthetized rats as a method for the production of experimental dysrhythmias and for the determination of infract size. J Pharmacol Methods 1980;3:357-68. Back to cited text no. 67 |
| 68. | Harris N, Kane KA, Muir AW, Winslow E. Influences of hypothermia, cold and isolation stress on the severity of coronary artery ligation induced arrhythmias in rats. J Pharmacol Methods 1982;7:161-71. Back to cited text no. 68 |
| 69. | Bernier M, Hearse DJ, Manning AS. Reperfusion induced arrhythmias and oxygen derived free redicals studies with "anti free redical" interventions and a free redical generating system in the isolated perfused rat heart. Cir Res 1986;58:331-40. Back to cited text no. 69 |
| 70. | Leparn I, Koltai M, Siegmund W, Szekeres L. Coronary artery ligation, early arrhythmias and determination of the ischemic area in conscious rats. J Pharmacol Methods 1983;9:219-30. Back to cited text no. 70 |
| 71. | Inagaki K, Hahn HS, Dorn II GW, Rosen DM. Additive protection of the ischemic heart ex vivo by combined treatment with d-protein kinase C inhibitor and e-protein kinase C activator. Circulation 2003;108:869-75. Back to cited text no. 71 |
| 72. | Macleod BA, Moult M, Saint KM, Walker MJ. The antiarrhythmic efficacy of anipamil against occlusion and reperfusion arrhythmias. Br J Pharmacol 1989;98:1165-72. Back to cited text no. 72 |
| 73. | Black SC, Rodger IW. Methods for studying expermental myocardial ischaemia and reperfusion injury. J Pharmacol Toxicol Methods 1996;35:179-90. Back to cited text no. 73 |
| 74. | Black SC. In vivo models of myocardial ischaemia and reperfusion injury. Application to drug discovery and evaluation. J Pharmacol Toxicol Methods 2000;43:153-67. Back to cited text no. 74 |
| 75. | Chen J, Nagasawa Y, Zhu BM, Ohmori M, Harada K, Fujimura A, et al . Pravastatin prevents arrhythmias induced by coronary artery ischemia/reperfusion in anesthetized normocholesterolemic rats. J Pharmacol Sci 2003;93:87-94. Back to cited text no. 75 |
| 76. | Walker MJ, Curtis MJ, Hearse DJ, Campbell RW, Jause MJ, Yellon DM, et al . The Lambeth convention; guicelines for the study of arrhythmias in ischaemia, infarction and reperfusion. Cardiovass Res 1988;22:447-55. Back to cited text no. 76 |
| 77. | Capasso JM, Li P, Zhang X, Anversa P. Heterogenity of ventricular remodeling after acute myocardial infarction in rats. Am J Physiol 1992;262:486-95. Back to cited text no. 77 |
| 78. | Coronel R, Fiolet JW, Wilms-Schopman FJ, Schaapherder AF, Johnson TA, Gettes LS, et al . Distribution of extracellular potassium and its relation to electrophysiologic changes during acute myocardial ischemia in the isolated perfused porcine heart. Circulation 1988;77:1125-38. Back to cited text no. 78 |
| 79. | Linz W, Wiemer G, Scholkens BA. Beneficial effects of bradykinin on myocardial energy metabolism and infract size. Am J Cardiol 1997;80:118-23. Back to cited text no. 79 |
| 80. | Aaj A. Sudden death, ventricular fibrillation, ventricular defibrillation; historical review and recent advances. In: Aaj A, editor. Acute phase of ischemic heart disease and myocardial infarction. Boston, Mass: Matinus-Nijhoff; 1982. Back to cited text no. 80 |
| 81. | Parker KK, Lavelle JA, Taylor LK, Wang Z, Hansen DE. Stretch-induced ventricular arrhythmias during acute ischaemia and reperfusion. J Appl Physiol 2004;97:377-83. Back to cited text no. 81 |
| 82. | Kaplinskg E, Ogawa S, Balke CW, Derifus LS. Two periods of early ventricular arrhythmia in the canine acute myocardial infarction model. Circulation 1979;60:397-403. Back to cited text no. 82 |
| 83. | Wilde AA, Asknes G. Myocardial potassium loss and cell depolarisation in ischaemia and hypoxia. Cardiovasc Res 1995;29:1-15. Back to cited text no. 83 |
| 84. | Lukas A, Antzelevitch C. Phase 2 reentry as a mechanism of initiation of circus movement reentry in canine epicardium exposed to simulated ischemia. Cardiovasc Res 1996;32:593-603. Back to cited text no. 84 |
| 85. | Smith WT, Fleet WF, Johnson TA, Engle CL, Cascio WE. The Ib phase of ventricular arrhythmias in ischemic in situ procaine heart is related to changes in call-to-call electrical coupling. Circulation 1995;92:3051-60. Back to cited text no. 85 |
| 86. | Barrett TC, MacLeod BA, Walker MJ. A model of myocardial ischaemia for the simultaneous assassment electrophysiological changes and arrhythmias in intact rabbits. J Pharmacol Toxicol Methods 1977;37:27-36. Back to cited text no. 86 |
| 87. | Oka J, Imamura M, Hatta E, Maruyama R, Isaka M, Murashita T, et al . Carrier-mediated norepinephrine release and reperfusion arrhythmias induced by protracted ischemia in isolated perfused guinea pig hearts: effect of presynaptic modulation by alpha(2)-adrenoceptor in mild hypothermic ischemia. J Pharmacol Exp Ther 2002;303:681-7. Back to cited text no. 87 |
| 88. | Premaratne S, Watanable BI, LaPenna WF, McNamara JJ. Effects of hyaluronidase on reducing myocardial infrac size in a baboon model of ischaemia reperfusion. J Surg Res 1995;58:205-10. Back to cited text no. 88 |
| 89. | Naslund U, Haggmark S, Johanson G, Pennert K, Peiz S, Marklund SL. Effects of reperfusion and superoxide dismutase on myocardial infract size in a closed chest pig model.Cardiovasc Res 1992;26:170-8. Back to cited text no. 89 |
| 90. | Billman GF. Role of ATP sensitive potassium channel in extracellular potassium accumulation and cardiac arrhythmias during myocardial ischaemia. Cardiovasc Res 1994;28:762-9. Back to cited text no. 90 |
| 91. | Aversane T, Ouryang P, Silverman H. Blockade of the ATP sensitive potassium channel modulates reactive hyperthermia in the canine coronary circulation. Circ Res 1991;69:618-22. Back to cited text no. 91 |
| 92. | Smith LL, Kukielka M, Billman GE. Heart rate recovery after exercise: A predictor of ventricular fibrillation susceptibility after myocardial infarction. Am J Physiol Heart Circ Physiol 2005;288:1763-9. Back to cited text no. 92 |
| 93. | Meizlish JL, Berger HJ, Plankey M, Errico D, Levy W, Zeret BL. Functional left ventricular aneurysm formation after acute anterior transmural myocardial infarction. N Engl J Med 1984;311:1001-6. Back to cited text no. 93 |
| 94. | Huang SK, Messer JV, Denes P. Significance of ventricular tachycardia in idiopathic dilated cardiomyopathy: observations in 35 patients. Am J Cardiol 1983;51:507-12. Back to cited text no. 94 |
| 95. | Hansen DE, Craig CS, Hondeghem LM. Stretch induced arrythmias in the isolated canine ventricle. Circulation 1990;81:1094-105. Back to cited text no. 95 |
| 96. | Hansen DE, Stacy Jr GP, Taylor LK, Jobe RL, Wang Z, Denton PK, et al . Calcium- and sodium-dependent modulation of stretch-induced arrhythmias in isolated canine ventricles. Am J Physiol Heart Circ Physiol 1995;68:1803-13. Back to cited text no. 96 |
| 97. | Cahn PS, Corvoni P. Current concepts and animal models of sudden cardiac death for the drug development. Drug Dev Res 1990;19:199-207. Back to cited text no. 97 |
| 98. | Chi L, Mu DX, Lucchesi BR. Electrophysiology and antiarrythmic actions of E-4031 in the experimental animal model of sudden coronary death. J Cardiovasc Pharmacol 1991;17:285-95. Back to cited text no. 98 |
| 99. | Krumpl G, Todt H, Schunder-Tatzver S, Raberger G. Holter monitoring in conscious dogs. Assesment of arrythmias occuring during ischaemia and in the early reperfusion phase. J Pharmacol Methods 1989;22:93-102. Back to cited text no. 99 |
| 100. | Takahara A, Hirasawa A, Dohmoto H, Shoji M, Yoshimoto R, Sugiyama A, et al . In vivo antiarrhythmic profile of AP-792 assessed in different canine arrhythmia models. Jpn J Pharmacol 2001;87:21-6. Back to cited text no. 100 |
| 101. | Dubray C, Boucher M, Paire M, Duchene-Marullaz P. A method for determining the atrial effective refractory period in the unanaesthetized dog. J Pharmacol Methods 1983;9:157-64. Back to cited text no. 101 |
| 102. | Paigen K. A miracle enough: The power of mice. Nat Med 1995;1:215-20. Back to cited text no. 102 |
| 103. | Lin MC, Rockman RA, Chin KR. Heart and lung disease in engineered mice. Nat Med 1995;1:749-51. Back to cited text no. 103 |
| 104. | 104.Charles IB, Christe ME, Aronovitz MJ, Seidman CE, Seidman JG, Mendelsohn ME. Electrophysiological abnormalities and arrhythmias in alpha MHC mutant familial hypertrophie cardiomyopathi mice. J Clin Invest 1997;99:570-6. Back to cited text no. 104 |
| 105. | Wu Y, Temple J, Zhang R, Dzhura I, Zhang W, Trimble R, et al . Calmodulin kinase II and arrhythmias in a mouse model of cardiac hypertrophy. Circulation 2002;106:1288-93. Back to cited text no. 105 |
| 106. | Charpentier F, Merot J, Riochet D, Le Marec H, Escande D. Adult KCNE 1- knockout mice exhibit a mild cardio cellular phenotype. Biochem Biophys Res Commun 1998;251:806-10. Back to cited text no. 106 |
| 107. | Berul CI, McConnell BK, Wakimoto H, Moskowitz IP, Maguire CT. Ventricular arrhythmia vulnerability in cardiomyopathic mice with homozygous mutant myosin binding protein C gene. Circulation 2001;104:2734-9. Back to cited text no. 107 |
| 108. | Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 1995;80:795-803. Back to cited text no. 108 |
| 109. | Wang Q, Curran ME, Splawski I, Burn TC, Millhalland JM, Van Raag TJ, et al . Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Net Genet 1996;12:17-23. Back to cited text no. 109 |
| 110. | Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the Ikr, Potassium channel. Cell 1995;81:299-307. Back to cited text no. 110 |
| 111. | Folco E, Mathur R, Mori Y, Buckett P, Koren G. A cellular model for long QT syndrome. J Biol Chem 1997;272:26505-10. Back to cited text no. 111 |
| 112. | London B, Jeron A, Zhou J, Buckket P, Han X, Mitchell GF, et al . Long QT and ventricular arrhythmias in transgenic mice expressing the N terminus and first transmembrane segment of a voltage-gates potassium channel. Proc Natt Acad Sci USA 1998;95:2926-31. Back to cited text no. 112 |
| 113. | Wakimoto H, Kasahara H, Tmaguire C, Izumo S, Berul CI. Coduction failure in mutant NKx 2.5 overexpression mice. Jan Card Elect 2002;13:682-8. Back to cited text no. 113 |
| 114. | Lande G, Demalombe S, Bammert A, Moormal A, Charpentier F, EsCande D. Transgenic mice overexpression human KvLQT1 dominant negative isoform. Part II: Pharmacological profile. Cardvasc Res 2001;50:328-24. Back to cited text no. 114 |
| 115. | London B, Baker LC, Lee JS, Shusterman V, Choi BR, Kubota T, et al . Calcium dependent arrhythmias in transgenic mice with heart failure. Am J Physiol Heart Circ Physiol 2003;284:431-41. Back to cited text no. 115 |
| 116. | Shinagawa K, Takeshita AS, Schram G, Nattel S. Effects of antiarrhythmic drugs on fibrillation in the remodeled atrium. Circulation 2003;107:1440-6. Back to cited text no. 116 |
| 117. | Zuanetti G, Corr PB. Antiarrhythmic efficacy of a new class III agent, UK-68, 798, during chronic myocardial infarction: evaluation using three dimensional mapping. J Pharmacol Exp Ther 1991;256:325-34. Back to cited text no. 117 |
| 118. | Task force of the working group on arrhythmias of the europian society of cardiology. The sicilian gambit. A new approach to the classification of antiarrhythmic drugs based on their actions on arrhythmogenic mechanism. Circulation 1991;84:1831-51. Back to cited text no. 118 |
| 119. | Bιnardeau A, Weissenburger J, Hondeghem L, Ertel EA. Effects of the t-type Ca2+ channel blocker mibefradil on repolarization of guinea pig, rabbit, dog, monkey, and human cardiac tissue. J Pharmacol Exp Ther 2000;292:561-75. Back to cited text no. 119 |
| 120. | Janse MJ, Wilms-Schopman F, Opthof T. Mechanisms of antifibrillatory action of Org 7797 in regionally ischemic pig heart. J Cardiovasc Pharmacol 1990;15: 633-43. Back to cited text no. 120 |
| 121. | Vermeulen JT, McGuire MA, Opthof T. Triggered activity and automaticity in ventricle trabeculae of failing human and rabbit heart. Cardiovasc Res 1994; 28:1547-54. Back to cited text no. 121 |
| 122. | Ypma JF. Adaption of refractory period of rat ventricle to changes in heart rate. Am J Physiol 1972;223:894-7. Back to cited text no. 122 |
| 123. | Krusche CA, Moller G, Beier HM, Adamski J. Expression and regulation of 17beta-hydroxysteroid dehydrogenase 7 in the rabbit. Mol Cell Endocrinol 2001;171:169-77. Back to cited text no. 123 |
Copyright 2005 - Indian Journal of Pharmacology
The following images related to this document are available:
Photo images
[ph05095t1.jpg]
|

{kind=link}