 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Indian Journal of Pharmacology, Vol. 38, No. 1, January-February, 2006, pp. 5-12 Education Forum Screening of antimalarial drugs: An overview Kalra BS, Chawla S, Gupta P, Valecha* N Department of Pharmacology, Maulana Azad Medical College, Bahadur Shah Zafar Marg, Delhi- 110002, *Malaria Research Center, No. 22, Sham Nath Marg
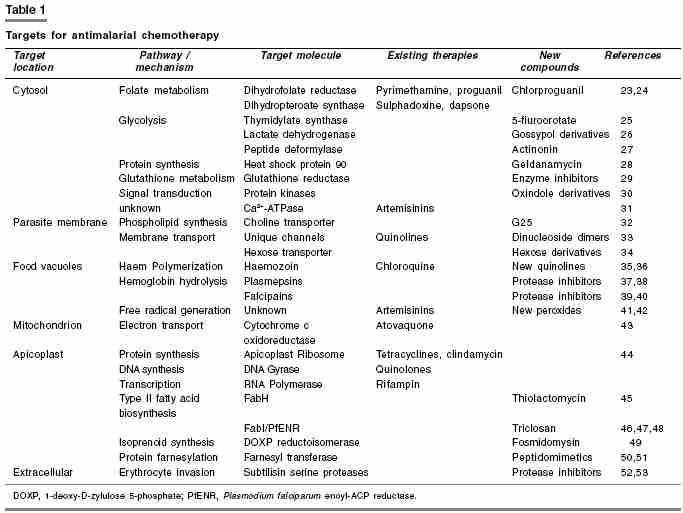
Delhi- 110054. Code Number: ph06002 Abstract Efforts to discover and develop new antimalarial drugs have increased dramatically in recent years mainly because of resistance to existing antimalarial drugs. Selection of candidate drugs for clinical trials in man and the design of clinical protocols are based upon consideration of data from a battery of preclinical test systems. All compounds are assessed initially in one or more primary screening models. A compound which is considered 'active' by well established criteria in primary screening test is considered for further evaluation in successively more clinical tests. At the end of each stage of testing, a decision is taken to advance the compound to the next stage or discontinue it. Primary screening tests should have optimal sensitivity, a high degree of reproducibility, high throughput, requiring a minimum quantity of test compound and should bear low cost. As there is growing need for newer and more efficacious antimalarial drugs especially in tropical countries, more sensitive and economical screening models are needed. This review is an update of various conventional and latest in vitro and in vivo screening methods being used for evaluation of antimalarial compounds.Keywords: Hypoxanthine uptake, rodent malaria model, Plasmodium berghei , primate model. Introduction Malaria is one of the oldest recorded disease in the world. In the 18th century the Italians associated malaria with ′bad air′- mala ria from where the name malaria is derived. It is a protozoal disease caused by parasites of the genus Plasmodium and transmitted to man by certain species of infected female Anopheline mosquito. Malaria remains one of the most important disease of developing world, killing 1-3 million people and causing disease in 300-500 million people annually worldwide.[1] In India, the National Malaria Eradication Programme started in 1958, achieved near complete disappearance of the disease in 1960s. However, due to development of insecticide resistance among mosquitoes and other factors, it staged a comeback in the 70s and continues to prevail in endemic or sub-endemic proportions in different regions. Clinically, malaria manifests as fever, chills, prostration and anemia. Severe form of the disease may lead on to delirium, metabolic acidosis, cerebral malaria, multi organ system failure, coma and death. Sporozoites inoculated with the bite of mosquito leads to development of blood stage infection (trophozoites) and gametocyte generation which are infectious for mosquito. Gametocytes in human blood are taken up by the mosquito leading to fertilization and zygote formation in mosquito midgut. This is followed by production of haploid sporozoite that invades the salivary glands of the mosquito and is subsequently transmitted back to humans due to the bite. Since last two decades, malaria control and treatment has been complicated by the emergence of resistance to widely used antimalarial drugs such as chloroquine. Drug resistance has been defined as the ability of parasite strain to multiply or survive in the presence of concentration of a drug that normally inhibit their multiplication or kill the parasite. To combat the problem of resistance, newer drugs are needed. Indeed, an unprecedented number of malaria discovery and development projects are now underway, involving many new drug targets for antimalarial therapy [Table - 1]. The goal is to develop safe and affordable drugs to counter the spread of malaria parasite resistant to existing drugs. This article is an endeavor in providing hands on information to post graduate and research students about various in vitro and in vivo screening methods being followed or recommended for antimalarial drug development. In vitro methods for screening antimalarial compounds Plasmodium falciparum can now be maintained in continuous culture in human erythrocytes incubated at 380C in RMPI 1640 medium with human serum or albumax (a lipid rich bovine serum albumin). Albumax appears to reduce both the rate at which erythrocytes deteriorate in vitro as well as pH drift when cultures are exposed to ambient air. Continuous culture was made possible by the observation that parasites develop better in a settled layer of red cells with a continuous slow flow of medium over it. In this method, a suspension of human AB group erythrocytes is inoculated with a small amount of falciparum infected Aotus monkey blood. Type AB blood is used because it can be mixed with Aotus blood without danger of agglutination of Aotus cells. This suspension is placed in flow vials, which provide flow of medium at 2 ml/hr over the settled cells and an atmosphere of 7% CO2 and 5% O2. The cultures are diluted with fresh human red cells on the fourth day and then every third or fourth day as growth continues.[2] Depending on the cell line selected, parasites propagate 3-8 fold every 48 h, thus care must be taken to avoid parasite cultures attaining too high a parasitemia (i.e. percentage of parasitized erythrocytes). Most lines grow optimally at 0.5-4% parasitemia. Parasites are most suitable for drug assays when there is 2-5% parasitemia with mostly ring stages and few or no gametocytes.[3] All stages of the erythrocytic cycle of parasite are present in the culture. Infectivity can be demonstrated by the inoculation of culture material intravenously into splenectomized Aotus monkey. Culture of Plasmodium falciparum is now being used to study the mode of entry of parasite into erythrocytes, screening of new drugs, to isolate and characterize strains and clones, to identify immunogenic antigens and genome of parasite. Several well characterized strains can be made available, either from academic laboratories[4] or through website www.malaria.mr4.org Materials and Methods 3H Hypoxanthine uptake
Percent reductions are used to plot percentage inhibition of growth as a function of drug concentration. IC[50] are determined by linear regression analyses on the linear segments of the dose response curve.[4] It is the most commonly used method for assessing antimalarial efficacy of a compound in vitro. Its shortcomings are, the method is expensive, complicated and involves usage of radioactive substance. Giemsa stained slide method (MIC method) In this model, parasites are incubated in a 5% suspension of erythrocytes with an initial parasite density of 1-2% at 370C. A sealed incubation chamber, continuously gassed with a mixture of 2% O2, 8% CO2, 90% N2 is used. Increase in the proportion of infected RBCs is assessed at the end of 72 hr incubation period in control samples and at various concentrations of each drug. This method relies on a morphological criterion of response and reports a single concentration as the end point i.e. concentration of a drug in the first sample showing complete inhibition of growth. This measurement is classically known as the Minimum Inhibitory Concentration (MIC), method which is suitable for distinguishing susceptible and resistant isolates.[5]
Other in vitro methods Isobologram analysis Micro-test (Mark III) Chloroquine : 1 - 64 *all drug concentrations are expressed as pmol per well Patient′s blood sample is inoculated in the wells and incubated with suitable medium. The number of schizonts with 3 or more nuclei out of a total of 200 asexual parasites is counted and compared with control well. For monitoring the level and spread of resistance, molecular diagnostic methods for detecting resistant parasite have been proposed.[11] These methods are suitable for use on a large number of samples in malaria endemic areas and have major advantage over in vitro tests that require parasite cultivation which take days to perform.[12] These molecular tools are based on the detection by PCR of point mutation in the parasite genome responsible for in vitro resistance. Advantages of in vitro methods
Limitations of in vitro methods
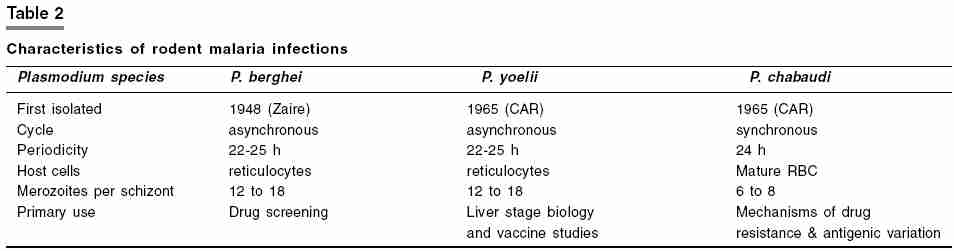
In vivo methods for screening antimalarial compounds Rodent models: a) Plasmodium berghei 4 day suppression test: On day 1 to 3, the experimental groups are treated again (with the same dose and same route) as on day 0. On day 4, 24 h after the last dose (i.e. 96 h post-infection), blood smears from all animals are prepared with Giemsa stain. Parasitemia is determined microscopically by counting 4 fields of approximately 100 erythrocytes per field. For low parasitemias (< 1%), up to 4000 erythrocytes have to be counted. The difference between the mean value of the control group (taken as 100%) and those of the experimental groups is calculated and expressed as percent reduction or activity using the following equation:
For slow acting drugs, additional smears should be taken on days 5 and 6, to determine parasitemia from which the activity is calculated accordingly. Untreated control mice typically die approximately one week after infection. For treated mice the survival-time (in days) is recorded and the mean survival time is calculated in comparison with the untreated and standard drug treated groups. Mice without parasitemia on day 30 of post-infection are considered cured. Compounds identified as active in this test are progressed through several of the following secondary tests. In the ′ Dose ranging full four day test ′, compounds are tested at four doses, by different routes of administration, to determine ED[50] value by plotting the log dose against probit activity. This test also leads to information about relative potency and bioavailability. In the ′ onset/recrudescence test′ , mice are administered with single dose of the test compound on day 3 of postinfection, subcutaneously. Control mice receive the suspension vehicle alone. Blood smears are prepared at intervals of 12 h, 24h and then daily till day 33, Giemsa stained and assessed for parasitemia. Results are expressed in terms of rapidity of onset of activity, time to onset of recrudescence, increase of parasitemia and duration of survival (in days). Compounds are also tested for prophylactic activity by administrating them prior to infection, followed by daily examination of smears. b) Hill′s test for causal prophylaxis and residual activity:[13] Phase 1. This basic procedure involves detection of causal prophylactic activity of the test compound in mice. Test compound is administered 3hr after a sporozoite inoculation. During the ensuing 14 day period, blood films are taken until 2% parasitemia is achieved. If parasitemia is not detected for 14 days the compound is considered to be fully protective. Phase 2 . Compound is then tested for residual activity directed against blood stage parasites by administrating a single dose of the test compound 48hr before 104 trophozoites are injected intravenously. If the time interval to reach 2% parasitemia is similar to that of the control group, then it is considered that no residual activity has occurred. Phase 3 . Compounds suspected of prolonged residual activity are tested by administering sporozoites followed by the drug 3 h later. After an additional 48h have elapsed, 0.2 ml blood is removed from each mouse and injected intraperitoneally into a clean mouse. Blood films from recipient mice are examined for a 14 day period or until patency develops. Residual activity is noted if less than 50% of the recipients develop parasitemia. A compound has no residual activity if 75% or more recipient mice develop patent infection. Phase 4. An additional procedure is also done to clarify whether or not a compound has residual effect on erythrocytic stages during the 48hr period of drug exposure in vivo . If no residual effect is seen then these parasites remain infective. To determine if the erythrocytic parasites are still viable, mice are injected intravenously with 104 trophozoites 48 h after the compound administration. After an additional 3-4 h, 0.2ml of blood is removed and injected to clean recipient mice. Blood films are taken and a comparison of the time interval to reach 2% parasitemia is made with control mice, if the time interval is similar, it reflects that no permanent damage has been done to the parasites and thus no residual activity is present. Sporonoicidal activity testing On the seventh day after the blood feed, a sample of each batch is removed and dissected. Midguts are examined with the aid of semi-dark ground illumination and oocyst counts are made. The mean count for each batch is calculated. To determine the inhibitory effects of a drug on oocyst development, it is dissolved in 4% sucrose and fed ad libitum to the insects following the blood meal. Usually a batch of 10 surviving A. stephensi for each drug concentration and for each control is sufficient to provide data from which the level of drug activity can be calculated from a comparison of mean oocyst counts in treated and control batches. A new in vivo mice model for antimalarial efficacy is recently designed which comprises of use of immunocompromised mice.[16] It can support Plasmodium falciparum infection as it lacks T and LAK ( Lymphokine activated killer cell ) cells. Immunodeficient mice have been widely used as xenogenic transplantation models allowing in vivo investigation of human cells and organs. In this model, P. falciparum parasitized human red blood cells ( P. falciparum -huRBC) can be grafted into immunodeficient BXN laboratory mice (P. falciparum -huRBC-BXN). For immunomodulation dichloromethylene diphosphate (Cl2MDP) is administered and is responsible for reducing tissue macrophages. The increase in polymorphonuclear neutrophils which occurs with parasitic infection is controlled by using an NIMP-R 14 monoclonal antibody. This is the first rodent model in which Plasmodium falciparum infection can be maintained. Test drugs are administered when parasitemia becomes stable for 6 days and smears show predominance of ring forms. Avian models Over time immemorial there have been many attempts to treat malarial fever with the wearing of charms and bracelets. It is interesting to note that Baranger and Filer (1953) using rings, bands or spirals of various metals placed around chicks infected with P. gallinaceum found some protective effects.[17],[18] Not only was there a prolonged survival time when compared with the untreated birds, but the asexual blood parasites were also retarded. Even more intriguing was that the removal of the band was followed by an increase in parasitemia on the following day! Copper, iron or gold were the most effective. Method: Direct inoculation of infected blood is usually employed for routine maintenance or to inoculate a number of animals for chemotherapy experiments. When only one or two birds need to be infected, a sufficient quantity of parasitized blood can be obtained from the leg vein or wing vein of an infected bird. When large number of chicks is to be infected from a single donor, blood is aspirated directly from the neck vein or from the heart. The total number of parasitized erythrocytes used to infect experimental animals is dependent on virulence of the strain and the route of inoculation. For production of slow infections which takes around 7 days to become patent, parasitized erythrocytes are injected into peritoneal cavity directly below the sternum, intramuscularly into the breast muscle, or subcutaneously into the neck region. For production of fast, easy and uniform infection, parasitized erythrocytes are injected intravenously into the neck vein; this fast infection leads to death in 5 to 7 days. Drugs may be administered to chicks orally, intravenously or intramuscularly, however, the oral route is the method used more frequently. Experimental birds are infected intravenously and receive the first dose of drug on day 1 followed by twice daily for the next 3 days. Blood films are made on day 4. Parasitemia achieved on day 4 in treated and untreated control group is then compared. Avian models have become unpopular primarily due to the introduction of the rodent models, as mammalian models are more relevant to human infections. Primate models Utility of primate models
These primate hosts are primarily used for screening of antimalarial drugs. They also serve as faithful models to investigate various complications associated with malaria. Aotus is one of the WHO recommended model for studies in malaria, and these are the only models which can sustain malarial infection caused by Plasmodium falciparum and P. vivax. Plasmodium cynomolgi rhesus model The animals receive 7 daily oral doses of the drug and blood films are examined daily during and after therapy up to a total of 30 days. If still negative, the animals are then examined twice weekly until they relapse. If the parasites are not cleared by the drug during the primary attack, the animals are given 7 daily doses of 5mg/kg of chloroquine. If no relapse follows chloroquine administration, it indicates that the test drug has destroyed the hypnozoites. When no relapse occurs within 8-12 weeks, the animals are splenectomized. Failure to develop further parasitemia within 4 weeks of this procedure indicates that the animals are radically cured since, in 99% of those that are not splenectomized, the infection relapses within 3 weeks of splenectomy. Since last 10 years, considerable progress has occured in the development of malarial vaccine. More than 40 distinct antigens in the various cycle stages of the parasite have been proposed as potential vaccine candidates. Owl and squirrel monkeys are excellent models for testing vaccines. Various studies have shown that there are three readily apparent advantages of monkey trials over human trials with proposed malarial vaccine: they provide efficacy data before clinical grade production; they might prevent the need for existing field trials; they might provide the data to validate in vitro assays.[20] The Clinical Trials Center at the Walter Reed Army Institute of Research, Silver Spring is involved with ongoing Challenge study. The Challenge study is being conducted to determine safety and immune response of candidate malarial vaccine.[21] After vaccination, it is important to see whether the vaccines are able to protect people against malaria. In order to do this, volunteers are infected with a fully drug-sensitive strain of malaria. In addition to vaccinated volunteers, a number of unvaccinated controls take part in the Challenge study to make sure that the infection system works and to act as a comparison. Volunteers are exposed to the bites of five malaria-infected mosquitoes. These mosquitoes are placed in paper cups onto which the volunteers rest their arm for five minutes. Untreated malaria could be very serious and so the volunteers are followed-up very carefully (up to twice per day). At each of these visits a blood sample is taken and examined for malaria. At the first sign of malaria, the volunteer is immediately treated with the antimalarial drug chloroquine. Volunteers getting full protection from the malaria vaccine would not develop the disease. The aim of the malaria Challenge study is to establish whether vaccination regimes offer protection against malaria infection. Protection may be ′partial′ or ′complete′: Complete protection is where vaccinated volunteers do not develop malaria in the Challenge study. Naturally, all the unvaccinated control volunteers develop malaria so that it becomes very clear that the infection system has worked. Partial protection is when there is a delay in the onset of malaria in the vaccinated volunteers compared to the unvaccinated controls. This means that the body′s immune system is controlling the infection to start with but is ultimately overwhelmed. Malaria vaccine development is a very difficult and challenging task due to various reasons. First, the size and genetic complexity of the parasite presents thousands of antigens to the human immune system. Due to antigenic diversity, identification of a useful target for vaccine development has been complicated, second, the parasite changes through several life stages even while in the human host, presenting a different subset of molecules for the immune system to combat with each stage. Third, the parasite has evolved a series of strategies that allow it to confuse, hide, and misdirect the human immune system. Finally, it is possible to have multiple malaria infections of not only different species but also of different strains at the same time.[22] Conclusion Malaria models have been established in a variety of laboratory animals, short and long term plasmodium culture systems have been extensively used for chemotherapeutic studies. A variety of avian, rodent, simian and human plasmodium models are used for screening and evaluation of candidate antimalarial compounds. The choice of malaria model depends upon sensitivity, reproducibility and breadth of response to known antimalarial drug and also on practical consideration such as required rate of testing, technical complexity, quantity of test compound needed and cost per test. Host factors such as natural resistance and immune competence also influence the efficiency of test compounds in different models. Each malaria model system has its individual characteristics, and no single model exists which can be said to be entirely predictive for humans[53].References
Copyright 2006 - Indian Journal of Pharmacology The following images related to this document are available:Photo images[ph06002f1.jpg] [ph06002t2.jpg] [ph06002t1.jpg] |
| |||||||||



{kind=link}
{kind=link}
{kind=link}