 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
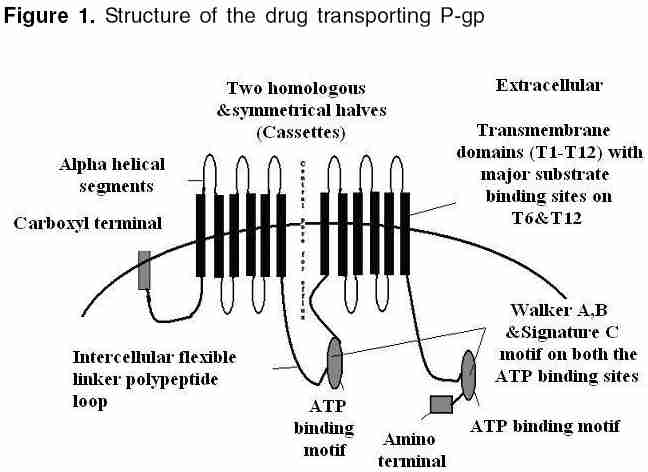
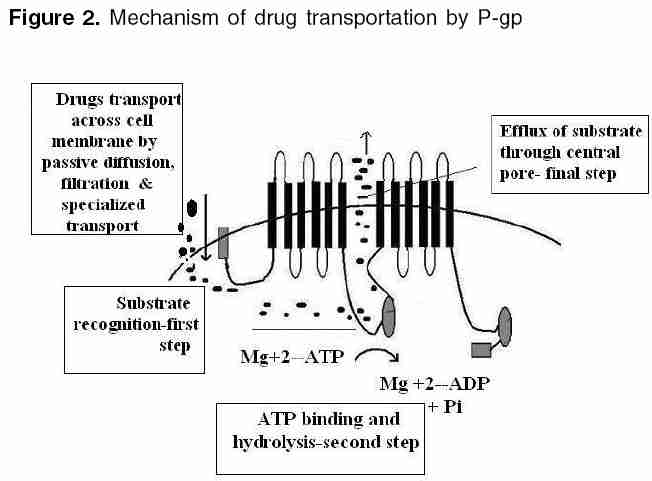
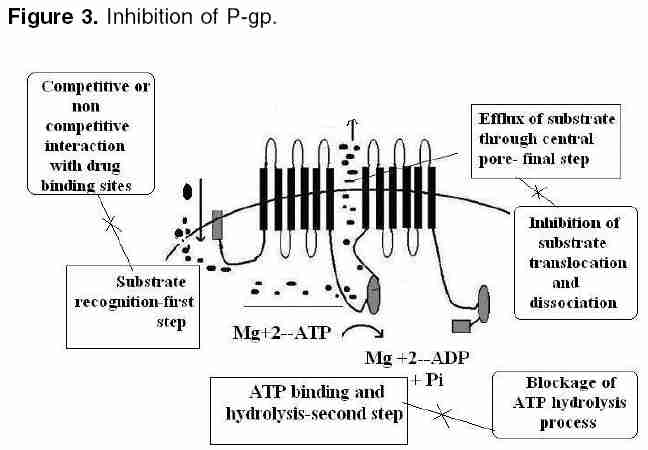
Indian Journal of Pharmacology, Vol. 38, No. 1, January-February, 2006, pp. 13-24 Education Forum P-glycoprotein: Pharmacological relevance Tandon Vishal R, Kapoor B, Bano G, Gupta S, Gillani Z, Gupta S, Kour D Department of Pharmacology & Therapeutics, Govt. Medical College, Jammu 180001, India Code Number: ph06003 Abstract P-glycoprotein (P-gp) is a 170 kDa membrane-bound protein, an energy-dependent efflux transporter driven by ATP hydrolysis. It is responsible for multidrug resistance of many drugs. Physiologically, it is involved in limiting the harmful exposure of toxins, drugs, and xenobiotics to the body by extruding them out of cells. It is increasingly recognized to play an important modulating role in the pharmacokinetic properties of many clinically important therapeutic agents and because of its importance in pharmacokinetics, its screening has to be incorporated into the drug discovery process. The modulation of drug transporters through inhibition or induction by various drugs or herbs can lead to significant drug-drug or drug-herb interactions by affecting various pharmacokinetic parameters of the drug. In addition, genetic polymorphism of P-gp has also been reported, which may affect drug disposition, produce variable drug effects, and may change disease risk susceptibility. As drug interactions and genetic polymorphism are important factors to be considered during drug development, P-gp may have an impact on drug development in future.Keywords: ABC transporter, multiple drug resistance-1, P-gp Introduction P-glycoprotein (P-gp) belongs to ABCB (MDR/TAP) superfamily of ATP-binding cassette (ABC) transporters. ABCB1 is also known as multiple drug resistance-1 (MDR1) or P-gp. It is the first human ABC transporter cloned and characterized through its ability to confer a MDR phenotype to cancer cells. The ABC genes represent the largest family of transmembrane (TM) proteins. These proteins bind ATP and use the energy to drive the transport of various molecules across all the cell membranes. ABC transporter superfamily is located in the plasma membrane or in intracellular membranes. In humans, 49 ABC transporters have been identified.[1], [2] P-gp was originally recognized as a surface glycoprotein, which was overexpressed in drug-resistant Chinese hamster ovary cell mutants by Juliano and Ling in 1976.[3] In humans, two members of the P-gp gene family (MDR1 and MDR3) exist, whereas three members of this family ( mdr1a, mdr1b0 and mdr20 ) are found in animals.[4],[5] The human MDR1/P-gp extrudes a variety of drugs across the plasma membrane and is widely distributed, whereas the homologous MDR3/P-gp has a more restricted expression with highest expression in liver in the canalicular membranes of hepatocytes and is required for phosphatidylcholine secretion into bile.[6] However, the involvement of human MDR3/P-gp in drug transport has been recently reported. The rate of MDR3/ P-gp-mediated transport is, however, low for most drugs, explaining why this protein is not detectably involved in multidrug resistance or carry much pharmacological relevance.[7] The human MDR1 / P-gp is ubiquitously expressed and is probably one of the most important ABC transporters for drug disposition in humans and it carries great pharmacological relevance. It has been implicated as a primary cause of MDR. The P-gp efflux transporter can functionally protect the body against toxic xenobiotics and drugs by excreting these compounds into bile, urine, and the intestinal lumen and by preventing their accumulation in brain, gonads, and placenta. It carries great pharmacokinetic importance and is significantly involved in many drug interactions.[4],[5] Thus, an attempt is made to briefly summarize the current knowledge of P-gp and to understand its structure, mechanism, and pathophysiological and pharmacokinetic roles in the present review. Cellular localization of (MDR1)/ P-gp In normal human tissues it is found to be concentrated in a small number of specific sites. Certain cell types in liver, pancreas, kidney, colon, and jejunum showed specific localization of P-gp. In liver, P-gp is found exclusively on the biliary canalicular front of hepatocytes and on the apical surface of epithelial cells in small biliary ductules. In pancreas , P-gp is found on the apical surface of the epithelial cells of small ductules but not larger pancreatic ducts. In kidney , P-gp is found concentrated on the apical surface of epithelial cells of the proximal tubules. Both colon and jejunum show high levels of P-gp on the apical surfaces of superficial columnar epithelial cells. Adrenal gland shows high levels of P-gp, diffusely distributed on the surface of cells in both the cortex and medulla.[9] Its expression is also detected in specialized epithelial cells with secretory or excretory functions, trophoblasts in the placenta , and on endothelial cells of capillary blood vessels at blood-tissue barrier sites.[8] In addition, it is found in the gastrointestinal epithelium, epithelia of the bronchi, mammary gland, prostate gland, salivary gland, and sweat glands of the skin.[10] Even, it is expressed in the human fetus, suggesting that it has a pivotal role in physiology of various organs already in early phases of human development.[11] [Figure - 1] shows that P-gp is a 170 kDa membrane-bound protein, an energy-dependent efflux transporter driven by ATP hydrolysis. It is composed of two homologous and symmetrical halves (cassettes), each of which contains six TM domains that are separated by an intracellular flexible linker polypeptide loop , approximately 75 amino acids in length with an ATP-binding motif.[12] The flexible secondary structure of the connector region is sufficient for the coordinate functioning of the two halves of P-gp, which is likely required for the proper interaction of the two ATP-binding sites.[13] There are two ATP-binding domains of P-gp, located in the cytosol side. ATP-binding domain(s) are also known as nucleotide-binding folds (NBFs). The NBFs are located in the cytoplasm and they transfer the energy to transport the substrates across the membranes. ABC pumps are mostly unidirectional. Each ATP-binding domain contains three regions: Walker A, B, and signature C motifs. Highly conserved Lys residue within the walker A motif of histadine permease is directly involved with the binding of ATP[14] and a highly conserved Asp residue within the walker B motif serves to bind the Mg[2]+ ion. Human P-gp, the MDR1 gene product, requires both Mg([2]+)-ATP-binding and hydrolysis to function as a drug transporter. It has also been proposed that magnesium may play a role in stabilizing the ATP-binding site.[15] Signature C motifs probably participate to accelerate ATP hydrolysis via chemical transition state interaction[16] and is also suggested to be involved in the transduction of the energy of ATP hydrolysis to the conformational changes in the membrane-integral domains required for translocation of the substrate.[17] Unlike the ATP-binding sites that are restricted to Walker A motifs of ATP-binding domains, many substrate-binding sites have been identified throughout the transmembrane (TM) domain of P-gp. The major drug-binding sites reside in or near TM6 and TM12.[18] In addition to this, TM1, TM4, TM10, and TM11 have drug-binding sites.[19] Amino acids in TM1 are involved in the formation of a binding pocket that plays a role in determining the suitable substrate size for P-gp, whereas Gly residues in TMs 2 and 3 are important in determining substrate specificity. The close proximity of TM2/TM11 and TM5/TM8 indicates that these regions between the two halves must enclose the drug-binding pocket at the cytoplasmic side of P-gp. They may form the "hinges" required for conformational changes during the transport cycle.[20] In addition to the TM domains, intracellular loops and even ATP-binding domains have drug-binding sites.[4] Each of the two TM domains of P-gp consists of six long α-helical segments. Five of the α-helices from each TM domain are related by a pseudo-twofold symmetry, whereas the sixth breaks the symmetry. The two α-helices positioned closest to the (pseudo)symmetry axis at the center of the molecule appear to be kinked.[21] P-gp has amino-and carboxyl-terminals. Initially, it was believed that N-terminal ATP-binding domain contains all residues necessary to hydrolyze ATP without interacting with the C-terminal ATP-binding domain.[22] But now it is believed that both the amino- and carboxyl-terminal ATP sites can hydrolyze ATP. However, there is no evidence that ATP can be hydrolyzed simultaneously by both sites.[23] [Figure - 2] depicts that drugs or substrates can cross into the cell membrane by simple diffusion, filtration, or by specialized transport, and the first step in drug efflux is drug recognition by P-gp followed by ATP-binding and subsequent hydrolysis. Finally, the generated energy is utilized to efflux substrate outside the cell membrane through central pore. The details of various steps are as follows. Drugs/substrate recognition: The major drug binding sites reside in or near TM6, TM12, TM1, TM4, TM10, and TM11.[18],[19] Amino acids in TM1 are involved in the formation of a binding pocket that plays a role in determining the suitable substrate/drug size for P-gp, whereas Gly residues in TM2 and TM3 are important in determining substrate specificity. Hence, these transmembrane domains help in large to recognize substrates/drugs. ATP-binding and subsequent hydrolysis: It is clear that ATP-binding and subsequent hydrolysis are essential for drug transport.[24] Around 0.6-3 molecules of ATP are hydrolyzed for every molecule of the drug transported outside the cell.[24] On the other hand, the study by Sauna and Ambudkar[25] suggested that two ATP molecules are hydrolyzed for the transport of every substrate molecule and demonstrated two distinct roles for ATP hydrolysis in a single turnover of the catalytic cycle of P-gp, one in the transport of substrate and the other in effecting conformational changes to reset the pump for the next catalytic cycle.[26] Detailed kinetic measurements have determined that both nucleotide-binding domains behave symmetrically and during individual hydrolysis events, the ATP sites are recruited in a random manner. Furthermore, only one nucleotide site hydrolyzes ATP at any given time, causing (in this site) a conformational change that drastically decreases (>30-fold) the affinity of the second site for ATP-binding. Thus, the blocking of ATP-binding to the second site although the first one is in catalytic conformation. This is referred as alternate catalytic cycle of ATP hydrolysis and ADP release is the rate-limiting step in the catalytic cycle and the substrates exert their effect by modulating ADP release.[27] Furthermore, it is proposed that the two nucleotide-binding domains dimerize to form an integrated single entity containing two-bound ATP with just one of the two ATP being hydrolyzed per dimerization event. If one ATP-binding domain is not functional, there is no ATP hydrolysis even when ATP binds to other ATP-binding domain.[28],[29] However, contrary to this, one study suggested that ATP hydrolysis by either one or both NBFs is essential to drive transport of solutes. Mutations of either NBF1 or NBF2 reduce solute transport, but do not abolish it completely.[30] Hence, common consensus still has to be made regarding ATP hydrolysis as one functional unit. Efflux of substrate/drug through central pore: P-gp intercepts lipophilic drugs as they move through the lipid membrane and flips the drugs from inner leaflets to the outer leaflet and to extra cellular medium.[31],[32] P-gp undergoes conformational changes on binding of nucleotide to the intracellular nucleotide-binding domains. Signature C motifs are probably suggested to be involved in the transduction of the energy of ATP hydrolysis to produce conformational changes in the membrane-integral domains required for translocation of the substrate.[17] The data reveal a major reorganization of the TM domains throughout the entire depth of the membrane on binding of nucleotide. On binding nucleotide, the TM domains reorganize into three compact domains that are each 2-3 nm in diameter and 5-6 nm deep. This reorganization opens the central pore along its length in a manner that could allow access of hydrophobic drugs (transport substrates) directly from the lipid bilayer to the central pore of the transporter.[31] However, recently, it has been proposed that drug substrates first diffuse from the lipid bilayer into the drug-binding pocket through "gates" formed by TM segments at either end of the drug-binding pocket and then effluxes the substrate through the central pore of the transporter to outside the membrane.[32] Substrates of P-gp: P-gp transports a wide range of substances with diverse chemical structure. In general, P-gp substrates appear to be lipophilic and amphiphatic.[4],[33] Among the drugs, important ones are enlisted below. Anticancer drugs: Actinomycin, cyclosporine-A, cisplatin, daunorubicin, docetaxel, doxorubicin, irinotecan, mitomycin-C, mitoxantrone, paclitaxel, teniposide, vinblastine, etoposide, imatinib and vincristine. Cardiovascular drugs: Atorvastatin, lovastatin, bunitrolol, celiprolol, talinolol, diltiazem, digoxin, digitoxin, losartan, quinidine, and verapamil. Antiviral drugs: amprenavir, indinavir, saquinavir, nelfinavir, and ritonavir Antibacterial agents: erythromycin, rifampin, sparfloxacin, levofloxacin, and pazufloxacin GIT drugs: Cimetidine, risperidone, domperidone, loperamide and ondansetron Others: Chloroquine, colchicines, dexamethasone, fexofenadine, morphine, phenytoin, tacrolimus, etc. [Figure - 3] depicts that inhibition of P-gp transport of a drug could potentially result from either competition for drug-binding sites without interrupting the ATP hydrolysis, such as itraconazole[34] and verapamil,[35] or it can result because of blockage of the ATP hydrolysis process, such as vandate,[26],[27] whereas drugs such as cyclosporine-A inhibit transport function by interfering with both substrate recognition and ATP hydrolysis.[4] Recently, another mechanism has been proposed for inhibition of P-gp-mediated drug transport by an allosteric mechanism . Unlike competitive inhibitors, cis-( Z )-flupentixol (a thioxanthene derivative) does not interfere with substrate recognition or ATP hydrolysis; instead, it prevents substrate translocation and dissociation, as a result of allosterical changes produced in drug transporter.[36] Inhibition can be partial , such as displacement of morphine binding by verapamil[37] or valeronitrile, a photoactive analog of verapamil inhibiting anthracyclines.[38] It can be complete like cyclosporine A, which can fully block the P-gp function in the blood-brain barrier (BBB) and the testes.[39] It can be competitive , suggesting that two substrates act on the same sites of P-gp and that only one or other can bind at any one time, for example, itraconazole, a known P-gp inhibitor displays competitive interaction with cimetidine.[34] On the other hand, digoxin and verapamil display noncompetitive interaction , meaning two substrates are able to bind simultaneously to P-gp molecule at distinct sites that are functionally independent.[35] Although recently, pyrrolopyrimidine [4] has been suggested to be a novel, potent, and selective MRP1 inhibitor with great selectivity against P-gp.[40] Pharmacological relevance of P-gp Multiple drug resistance Indeed, the overexpression of human and mouse MDR1/P-gp conferred resistance to multiple drugs such as vinca alkaloids, anthracyclines, epipodophyllotoxins, and taxols. Tumors arising from tissues where MDR1/P-gp is highly expressed show intrinsic resistance to chemotherapy, although acquired resistance is often correlated with an increased expression of MDR1/P-gp.[42] However, recent reports indicate that it is intercellular transfer of P-gp, which mediates acquired multidrug resistance in tumor cells and results in increased drug resistance both in vitro and in vivo .[43] P-gp induction by antineoplastics such as cisplatin, doxorubicin, or paclitaxel at concentrations related to the clinically achievable plasma peak concentration is one of the reasons for failure of ovarian cancer therapy.[44] Similarly, thymoma cell line EL4 cells and human CEM acute lymphoblastic leukemia cells express mRNA for multidrug resistance protein-1.[45],[46] Inhibition of P-gp represents a promising approach for treatment of multidrug-resistant tumors. Imatinib mesylate (signal transduction inhibitor) is a potent and selective tyrosine kinase inhibitor, which can effectively inhibit platelet-derived growth factor-induced glioblastoma cell growth preclinically. However, in patients, a limited penetration of imatinib into the brain by P-gp has been reported. Co-administration of P-gp inhibitors may improve delivery of imatinib to malignant gliomas.[47] Likewise, a series of 4-aryl-1,4-dihydropyridines and corresponding aromatized 4-arylpyridines have been synthesized to enhance multidrug resistance reversal activity successfully to restore intracellular accumulation of vinblastine in a resistant human breast adenocarcinoma cell line,[48] as well as polyene (-)-stipiamide has been designed and synthesized as new MDR reversal agents.[49] Not only this, researchers are developing now cytotoxic agents that are insensitive to P-gp-mediated drug efflux. Lamellarin D (LAM-D), a marine alkaloid endowed with potent cytotoxic activities against various tumor cells, in particular, human prostate cancer cells and leukemia cells, is one such addition. LAM-D acts as a potent proapoptotic agent and its cytotoxic action is fully maintained in multidrug-resistant cells compared with the sensitive parental cell line.[50] MDR1/P-gp not only causes multidrug resistance in cancer but has been found responsible for MDR of many other drugs. Anti-epileptic drug (AED) therapy: This is an unresolved problem in many patients with epilepsy, and mechanisms of intractability are not well understood. It has been hypothesized that overexpression of P-gp and other efflux transporters in the cerebrovascular endothelium in the region of the epileptic focus may lead to drug resistance in epilepsy. This hypothesis is supported by the findings of elevated expression of efflux transporters in epileptic foci in patients with drug-resistant epilepsy, induction of expression by seizures in animal models, and experimental evidence that some commonly used AEDs are substrates.[51] Brain entry of risperidone and 9-hydroxyrisperidone: similarly, the brain entry of risperidone and its active metabolite 9-hydroxyrisperidone is greatly limited by P-gp, resulting into failure of therapy in psychotics.[52] Chloroquine-resistant Plasmodium falciparum strains: These accumulate less drug than more sensitive parasites, and chloroquine resistance could be modulated in vitro by the classic MDR modulator verapamil.[53] The mechanism underpinning chloroquine resistance in the humans by malarial parasite P. falciparum remains controversial. By investigating the kinetics of chloroquine accumulation under varying trans conditions, recently, one study presented evidence for a saturable and energy-dependent chloroquine efflux system to be present in chloroquine-resistant P. falciparum strains. Verapamil and the calcium/calmodulin antagonist W7 abrogate are known inhibitors of P-gp-stimulated chloroquine accumulation,[54] whereas nonylphenolethoxylates, potential P. falciparum P-gp substrates and drug efflux inhibitors, can very effectively lead to reversal of mefloquine and quinine resistance in P. falciparum .[55] Induction of P-gp: This may be one of the mechanisms involved in the development of morphine tolerance. P-gp induction may enhance morphine efflux from the brain, thus reducing morphine′s pharmacological activity.[56] Drug resistance: This may also be induced by long-term treatment or high doses of prednisone as the result of P-gp (MDR-1) induction, and P-gp antagonists may improve the current therapeutic schemes for the treatment of myasthenia gravis.[57] Overexpression of multidrug transporters: this significantly reduces the accumulation of protease inhibitors (PIs) at major sites of virus replication, which potentially can accelerate the acquisition of viral resistance.[58] Even this has been suggested to play a role in antibiotic resistance, as evident from the fact that two novel mammalian MDR inhibitors, biricodar (VX-710) and timcodar (VX-853), are effective in lowering the MICs of several clinically used antibiotics, including fluoroquinolones.[59] In addition to this, P-gp-dependent reduced intracellular accumulation of fluconazole, antileishmania drugs, anthelmintic drugs, and multidrug resistance in the protozoan parasite, such as Entameba histolytica , [60],[61],[62],[63] is well documented. Hence, targeted inhibition of P-gp may represent an important strategy by which this problem of MDR can be overcome. Biological barrier to xenobiotic Transport mechanisms for the extrusion of toxic xenobiotics and their metabolites from cellular environment are crucial for living organisms. Accumulation of these toxins may affect a number of regulatory and other functions, ultimately leading to cell death. This trafficking of toxins and their metabolites is an energy-dependent primary active process involving the hydrolysis of nucleotide triphosphates (ATP), whereas transferring the substrates.[64] Therefore, the major physiological role of the multidrug transporters, especially P-gp, is the protection of our cells and tissues against xenobiotics.[65] 1,1-Bis(4-chlorophenyl)-2, 2, 2-trichloroethane (DDT) is an organochlorine pesticide. Its metabolite, 1,1-dichloro-2,2-bis( p -chlorophenyl)-ethene (p,p′-DDE), is a persistent environmental contaminant and both compounds accumulate in animals. DDT and p,p′-DDE′s ability to induce MDR1 gene function as a defence against xenobiotic exposure.[66] Similarly, MDR1/P-gp expression is severely increased by plastic-derived xenobiotics[67] and can decrease toxicity by removing environmental toxicants such as pesticides and heavy metals from cells in mammals. P-gp modulators should, therefore, be carefully used, because some modulators that reverse P-gp efflux action in vitro may lead to alterations of tissue function and increased toxicity of xenobiotics in normal tissues.[68] Blood brain barrier (BBB) also acts as a central nervous system (CNS)-detoxifying system for both endogenous substrates and xenobiotics in the brain, utilizing P-gp efflux mechanism predominantaly.[69] Even enhanced xenobiotic transporter expression in normal hepatocytes in response to environmental and chemotherapeutic toxins is well suggested, which may have protective role by limiting xenobiotic exposure to cells.[70] Pharmacokinetic implications of P-gp Absorption: More recently, the role of efflux transporters in determining the permeability and overall bioavailability of drugs has gained considerable attention. P-gp efflux pump is localized in a wide range of tissues, including enterocytes of the GI tract. An increasing number of drugs, including HIV protease inhibitors such as indinavir, ritonavir, and saquinavir, and anticancer drugs such as paclitaxel, docetaxel, as well as BMS-387032, a novel cyclin-dependent kinase 2 inhibitor, have been reported to be substrates for P-gp and it can significantly limit the oral bioavailability of these drugs in dose-dependent manner. [71],[72],[73] On the other hand, P-gp(s)-substrates such as etoposide, indinavir, ritonavir, saquinavir, and verapamil, which exhibited varying degrees of efflux, showed dose-independent in vivo kinetics in absorption ( Cmax and Tmax) and bioavailability (AUC).[74] On the other side, P-gp substrates, such as digoxin, paclitaxel, talinolol, and saquinavir, which exhibited high efflux, showed improved bioavailability in the presence of P-gp inhibitors. [75],[76],[77],[78] Thus, it should be appreciated that both passive permeability and the P-gp efflux process operating in mutually opposite directions, contribute to overall drug permeability and thus influence the bioavailability. Most of the P-gps are found towards the upper limits of molecular weight (>500) and calculated total polar surface area (>75 A2). This indicates that unfavorable chemical features of P-gps limit passive permeability, and thus are more susceptible to P-gp-mediated efflux.[79] Not only this, many important drug interactions by modulation of intestinal P-gp have been documented. Quinidine increases the absorption and plasma concentrations of oral morphine, suggesting that intestinal P-gp affects the absorption, bioavailability, and hence clinical effects of oral morphine.[80] Similarly, reducing effect on the saquinavir (SQV) oral bioavailability during ethanol consumption is owing to enhanced efflux of SQV at the intestine and liver, which is owing to excessive expression of P-gp caused by ethanol consumption.[81] Both increased oral bioavailability and reduced nonglomerular renal clearance of digoxin contribute to the interaction between digoxin and clarithromycin, probably owing to inhibition of intestinal and renal P-gp.[82] On the other hand, the increase in steady-state digoxin concentrations is produced by coadministration of 80 mg atorvastatin owing to inhibition of digoxin secretion into the intestinal lumen.[83] Rifampicin pretreatment also reduces plasma celiprolol (a β-adrenoceptor-blocking agent) concentrations, possibly by induction of P-gp, particularly in the intestinal wall, which leads to decreased absorption of celiprolol.[84] Drug distribution: BBB and placental barrier are very important determinants of drug distribution and P-gp is an important component of these biological barriers. Thus, it can influence the drug distribution of many therapeutic agents significantly. Blood brain barrier: The BBB contributes to brain homeostasis by protecting the brain from potentially harmful endogenous and exogenous substances. The P-gp has been demonstrated as a key element of the BBB that can actively transport a huge variety of lipophilic drugs out of the brain capillary endothelial cells that form the BBB. Consequences of P-gp efflux transporters in the BBB include minimizing or avoiding neurotoxic adverse effects of drugs that otherwise would penetrate into the brain. However, P-gp efflux transporters may also limit the central distribution of drugs that are beneficial to treat CNS diseases. Modulation of P-gp efflux transporters at the BBB forms a novel strategy to enhance the penetration of drugs into the brain and may yield new therapeutic options for drug-resistant CNS diseases.[85] P-gp can influence the concentration of many desired drugs in brain that are beneficial for the treatment of CNS disorders, such as, it may influence the intracerebral concentrations of nortriptyline (antidepressant drug),[86] risperidone,[52] and antiepileptic drugs.[51] Thus, in such situations, inhibition of P-gp is desirable. Modulation of P-gp in brain is possible by three possible ways: (i) direct inhibition by specific competitors, (ii) functional modulation, and (iii) transcriptional modulation. Each has the potential to specifically reduce P-gp function and thus selectively increase brain permeability of P-gp substrates.[87] Combined inhibitory effect of four P-gp inhibitors, cyclosporin A, verapamil, tetrandrine, and doxorubicin, has been suggested to be a more useful approach to increase drug concentration in brain tissues in order to enhance the therapeutic effect and to reduce the toxicity of drugs.[88] Similarly, amlodipine derivatives, CJX1 and CJX2, have been shown to possess a modulative effect on P-gp function in rat brain microvessel endothelial cells.[89] Chemical modification of the paclitaxel (Taxol) structure reduced its interactions with the product of the MDR1 gene, resulting in improved BBB permeability.[90] Another important function of P-gp in brain is to remove xenobiotics. The brain uptake of xenobiotics is restricted by the BBB formed by brain capillary endothelial cells. Active efflux transport systems in the BBB work as a detoxification system in the brain by facilitating removal of xenobiotic compounds from the brain.[91] BBB acts as a CNS-detoxifying system for both endogenous substrates and xenobiotics in the brain. Several transporters, most importantly P-gp in the brain capillary endothelial cells, appear to play an important role in reducing the brain level of hydrophilic endogenous substrates produced either in the brain or peripheral organs, e.g., neurotransmitters, neuromodulators, metabolites of neurotransmitters, and uremic toxins. It has been reported that large hydrophilic molecules, such as IgG, apotransferrin, and amyloid-β peptide, are also susceptible to brain-to-blood efflux transporter.[69] Its role in the etiology of some CNS diseases is also indicated. Overexpression of P-gp and other efflux transporters in the cerebrovascular endothelium, in the region of the epileptic focus, also may lead to drug resistance in epilepsy or intractable epilepsy.[51] A very interesting hypothesis has been postulated recently in relation to Parkinson′s disease (PD) and Alzheimer′s disease. The brain is normally protected from these noxious blood-borne chemicals by the BBB with the help of P-gp. PD and Alzheimer′s disease patients have reduced or dysfunctional P-gp function in the BBB, which may be one of the causative mechanisms for these states.[92],[93] Placental barrier: It has been shown that nearly all drugs that are administered during pregnancy will enter to some degree in the circulation of the fetus via passive diffusion. In addition, some drugs are pumped across the placenta by various active transporters located on both the fetal and maternal side of the trophoblast layer. It is only in recent years that the impact of active transporters such as P-gp on the disposition of drugs has been demonstrated.[94] Presence of P-gp in the rat chorioallantoic placenta starting soon after its development signifies the involvement of P-gp in transplacental pharmacokinetics during the whole period of placental maturing.[95] P-gp limits the entry of various potentially toxic drugs and xenobiotics into the fetus and is thus considered a placental protective mechanism. It is expressed on the brush-border membrane (maternal side) of human placental trophoblast cells. P-gp is considered to regulate the transfer of several substances including vinblastine, vincristine and digoxin from mother to fetus, and to protect the fetus from toxic substances.[96] Indinavir may be used in the management of HIV infection during pregnancy. Poor maternal-to-fetal transfer of indinavir has been reported, but the mechanisms remain unknown. Recently, involvement of placental P-gp has been suggested and the use of transport modulators can increase the maternal-to-fetal transfer of PIs.[97] P-gp appears to be involved in drug extrusion, in early preg nancy more than at term when the fetus is more susceptible to exposure of drugs and toxins.[98] One very contrary finding in placental drug disposition appeared that neither quinidine nor verapamil, known inhibitors of P-gp, affect the transplacental transfer of digoxin in vitro in normal human placenta. Thus, in contrast to the other tissues, they do not inhibit P-gp activity in term human placenta.[99] Its role in transplacental pharmacotherapy is also promising. Sustained fetal tachyarrhythmia is a potentially life-threatening condition for the unborn. Digoxin is commonly used as an initial monotherapy. The treatment success rate with digoxin is about 50%. Although transplacental pharmacotherapy has been available, it is a challenging task to maximize fetal drug exposure, whereas minimizing drug exposure of the mother. P-gp expressed in placenta decreases fetal exposure to maternal digoxin; thus, pharmacological manipulation of drug transporters may open a door to ultimate optimization of the transplacental pharmacotherapy.[100] Drug metabolism: Intestinal CYP3A4-mediated biotransformation and active efflux of absorbed drug by P-gp are major determinants of bioavailability of orally administered drugs. The hypothesis that CYP3A4 and P-gp may act in concert to limit oral drug bioavailability is attractive from a theoretical point of view. Evidence in support of such interplay between CYP3A4 and P-gp comes mainly from a limited number of in vitro and animal studies. Obviously, it is a challenging task to demonstrate in vivo in humans that the functions of CYP3A4 and P-gp in enterocytes are complementary and results to directly support this concept remain elusive. However, CYP3A4 and P-gp are clearly an integral part of an intestinal defense system to protect the body against harmful xenobiotics, and drugs that are substrates of both proteins often have a low bioavailability after oral administration.[101] The hypothesis that drugs which interact with the apical efflux pump P-gp may enhance CYP3A4-mediated disappearance of substrates was recently tested in many studies suggesting that P-gp/CYP3A4 are cosubstrates and that P-gp potentiates CYP3A4-mediated drug disappearance during intestinal secretary detoxification.[102] As well as it is possible that P-gp may influence first-pass metabolism in a co-operative manner.[103] Therefore, like CPY-mediated drug interactions, P-gp-mediated drug interactions may be anticipated when P-gp substrates and P-gp inhibitors or inducers are coadministrated. Because both proteins are expressed in enterocytes and hepatocytes and contribute to a major extent to first-pass elimination of many drugs, there is an overlapping of substrate specificities and inhibitor/inducers between CYP3A4 and P-gp; many drug interactions may involve both CYP3A4 and P-gp and it is difficult to distinguish the specific nature of drug interactions at present. P-gp inhibition: Pharmacokinetic parameters of paclitaxel are significantly affected by verapamil, which is an inhibitor of the metabolizing enzyme (CYP3A4) in the intestinal mucosa and liver, and the P-gp efflux pump in the intestinal mucosa.[104] Similarly, indinavir, a potent and specific inhibitor of HIV protease, is a known substrate of cytochrome P-450 (CYP) 3A and P-gp.[105] They may alter pharmacokinetics of each other. The low oral bioavailability of the saquinavir is dramatically increased by coadministration of ritonavir. Because saquinavir and ritonavir are substrates and inhibitors of both the drug transporter P-gp and of the metabolizing enzyme CYP3A4, the highly increased bioavailability of saquinavir because of ritonavir coadministration most likely results from reduced saquinavir metabolism.[106] Less potent P-gp inhibitors, such as valspodar (PSC-833), cyclosporin A, and ketoconazole, as well as quinidine and verapamil, had modest or little effect on brain-plasma ratios but increased plasma nelfinavir concentrations owing to inhibition of CYP3A-mediated metabolism.[107] Preliminary data indicate that piperine , a major component of black pepper, inhibits drug-metabolizing enzymes in rodents and increases plasma concentrations of several drugs, including P-gp substrates (phenytoin and rifampin) in humans. Piperine inhibits both the drug transporter P-gp and the major drug-metabolizing enzyme CYP3A4. Because both proteins are expressed in enterocytes and hepatocytes and contribute to a major extent to first-pass elimination of many drugs, therefore it could affect plasma concentrations of P-gp and CYP3A4 substrates in humans, in particular, if these drugs are administered orally.[108] Grapefruit juice components inhibit CYP3A4-mediated saquinavir metabolism and also modulate to a limited extent, P-gp mediated saquinavir transport in Caco-2 cell monolayer.[109] P-gp induction: P-gp and CYP3A have considerable overlap in inducers in vitro . Characterizing P-gp induction in vivo and potential coregulation with CYP3A are important goals for predicting drug interactions. Overall, P-gp induction in vivo is tissue-specific whereas, CYP3A induction is inducer-dependent.[110] The P-gp efflux pump can also influence the hepatocellular concentration of xenobiotics that are modulators and substrates of cytochrome P4503A (CYP3A).[111] Verapamil and propranolol, whose transepithelial permeability are unaffected by P-gp, appear to be effective inducers of P-gp expression in gut epithelial cells in vitro . On the other hand, the in vivo significance of these observations is unknown.[112] Extended exposure to the human immunodeficiency viral PI ritonavir (RIT) and dexamethasone (DEX) leads to increased expression and function of CYP3A isoforms in the liver and in enteric mucosal cells and on the expression of the efflux transport protein, P-gp in enteric mucosa, and in brain microvessel endothelial cells. Both RIT and DEX increase function and expression of enteric CYP3A, although to a more modest extent (about 1.7-fold for RIT, about 3.3-fold for DEX). Enteric P-gp expression was equally induced (by 2.8-fold) by both RIT and DEX,[113] suggesting them to be possible candidates for drug interactions. A chronic treatment with rifampicin induces the expression of transport proteins and of CYP 3A4 and could therefore alter kinetics of drugs that are their substrates.[114] Even phenobarbital can induce P-gp but it is unable to increase MDR-1 and MRP-1 protein levels significantly.[115] Similarly, doxorubicin can act as a potent acute inducer of functional P-gp.[116]Atazanavir is an inhibitor and inducer of P-gp as well as a potent inhibitor of CYP3A in vitro , suggesting a potential for atazanavir to cause drug-drug interactions in vivo .[117] St. John′s wort ( Hypericum perforatum ) is one of the most commonly used herbal antidepressants is a potent inducer of CYP 3A4 and P-gp thereby, affecting pharmacokinetics of many drugs.[118],[119] Drug excretion: Renal excretion - renal P-gp forms a transepithelial drug transport pathway that is responsible for the net urinary excretion of various xenobiotics and drugs in vivo. The renal excretion of a drug can essentially be divided schematically into three functional processes: glomerular filtration, tubular reabsorption and tubular secretion. Historically, two distinct tubular secretion mechanisms have been described for drugs: one via organic cations and the other via organic anions. More recently, a third tubular secretion mechanism has been identified, mediated by P-gp.[120] The synthetic surfactant, nonylphenol ethoxylate (NPE), is widely used and is a known hormone disrupter that is readily absorbed orally or topically. The current findings indicate that the function of kidney′s Mdr1p 0 may be to eliminate exogenous compounds from the body like NPE.[121] Numerous drug interactions can result, if P-gp in renal epithelium is modulated by some drugs. Reduced nonglomerular renal clearance of digoxin contributes to the interaction between digoxin and clarithromycin, probably owing to inhibition of intestinal and renal P-gp.[82] Digoxin, which has a very narrow therapeutic window, is one of the most commonly prescribed drugs in the treatment of congestive heart failure. In some cases of atrial fibrillation, digoxin is used in combination with verapamil. Verapamil can increase the plasma concentration of digoxin up to 60-90%. So far, the precise mechanism of this pharmacokinetic drug-drug interaction is not known. Many studies suggest that verapamil reduces the renal clearance of digoxin. Inhibition of P-gp activity by verapamil in noncompetitive manner may result in a decreased renal tubular elimination of digoxin.[35] Cyclosporine-A inhibits the transepithelial transport of digoxin mediated by human P-gp and therefore reduces the renal tubular secretion of digoxin by the kidney.[122] Pazufloxacin is excreted into the urine via P-gp. Cyclosporine significantly increases the steady-state concentration of pazufloxacin in plasma by decreasing the tubular secretion and glomerular filtration rate. Sparfloxacin, a P-gp substrate, also significantly decreases the renal and tubular secretion clearances of pazufloxacin, suggesting that pazufloxacin and sparfloxacin share the same transporters, including P-gp.[123] Similarly, the increased systemic exposure of cimetidine during coadministration with itraconazole is likely owing to inhibition of P-gp-mediated renal tubular secretion.[34] In chronic renal failure, the renal excretion of certain drugs is dramatically reduced. P-gp protein content and P-gp mRNA levels in chronic renal failure rats reduce with chronic renal failure, reflecting reduced intestinal drug elimination via a decrease in P-gp transport activity.[124] Biliary excretion: Effects of various types of P-gp modulation on the biliary excretion index (BEI; a relative measure of the extent of biliary excretion) and the in vitro biliary clearance, CL (bile) suggest significant reductions of BEI and CL (bile) in the presence of the P-gp inhibitors like verapamil and progesterone.The P-gp activator quercetin enhances CL (bile) by approximately fourfold, with only a minor effect on BEI, suggesting that quercetin has a more pronounced effect on uptake at the basolateral membrane rather than excretion across the canalicular membrane. Treatment of hepatocytes for 48 h with dexamethasone result in significant enhancement of CL (bile), whereas rifampin increases both BEI and CL(bile), indicating that inducing effects of dexamethasone and rifampin are occurring at the basolateral and canalicular membranes, respectively.[125] Azithromycin can modify the hepatobiliary excretion of doxorubicin-a substrate for P-gp in vivo .[126] Doxorubicin, cyclosporine A, and erythromycin can significantly decrease the biliary clearance of grepafloxacin (GPFX). Cyclosporine A and erythromycin have a much stronger inhibitory effect on the biliary excretion of GPFX than doxorubicin[127] whereas; erythromycin competitively inhibits P-gp-mediated biliary excretion of doxorubicin.[128] All these findings suggest that P-gp plays a very important role in biliary drug excretion and various drug interactions are possible. Herbal drug modulation: Many herbal dugs have been suggested to produce drug interaction mediated by P-gp modulation. St. John′s wort ( H. perforatum ) is one of the most commonly used herbal antidepressants. St. John′s wort decreases the blood concentrations of amitriptyline, cyclosporine, digoxin, fexofenadine, indinavir, methadone, midazolam, nevirapine, phenprocoumon, simvastatin, tacrolimus, theophylline, and warfarin. It decreases the plasma concentration of the active metabolite SN-38 in cancer patients receiving irinotecan treatment. St. John′s wort did not alter the pharmacokinetics of tolbutamide, but increased the incidence of hypoglycemia. Several cases have been reported on the fact that St. John′s wort decreases cyclosporine blood concentration, leading to organ rejection. St. John′s wort caused breakthrough bleeding and unplanned pregnancies when used concomitantly with oral contraceptives. It also causes serotonin syndrome when coadministered with selective serotonin-reuptake inhibitors, e.g., sertaline and paroxetine. This happens because it is a potent inducer of CYP 3A4 and P-gp, although it may inhibit or induce other CYPs, depending on the dose, route, and duration of administration.[118],[119] On the other hand, piperine, a major component of black pepper, inhibits human P-gp and/or CYP3A4 and can affect plasma concentrations of P-gp and CYP3A4 substrates in humans.[108] Similarly, grapefruit juice components inhibit CYP3A4-mediated saquinavir metabolism and also modulate, to a limited extent, P-gp-mediated saquinavir transport.[109] Bitter melon fraction might be involved in the drug bioavailability by specifically inhibiting the efflux mediated by P-gp.[129] Dietary phytochemicals, such as capsaicin, curcumin, [6]-gingerol, and resveratrol, have inhibitory effects on P-gp and have potencies to cause drug-food interactions.[130] Curcumin-I is the most effective MDR modulator among curcuminoids and may be used in combination with conventional chemotherapeutic drugs to reverse MDR in cancer cells.[131] The organosulfur compounds present in a garlic-rich diet might alter chemotherapeutic treatments using P-gp or Mrp2 0 substrates.[132] Elevation of P-gp function by a catechin in green tea is also reported.[133] Coadministration of fexofenadine with orange or apple juice to rats decreases the oral exposure of fexofenadine by 31% and 22%, respectively, mediated by P-gp.[134] P-gp polymorphism: In addition to physiological and environmental factors, expression and function of P-gp are modified by genetic polymorphisms of the MDR1 gene. So far, several MDR1 single-nucleotide polymorphisms have been identified, and mutations at positions 2677 and 3435 have been associated with alteration of P-gp expression and/or function. In contrast with drug-metabolizing enzymes (e.g., CYP2D6), for which loss of function mutations or gene amplification manifests as distinct phenotypes in the population, the impact of MDR1 polymorphisms on pharmacokinetics and pharmacodynamics of P-gp substrates is moderate. MDR1 polymorphisms may affect drug disposition, can result into variable drug effects, and can change disease risk susceptibility.[4],[33],[135] Intracellular concentration of HIV PIs and antiretroviral efficacy are affected by variable P-gp expression, as a result of the polymorphism of MDR1 3435 C/T.[136] Phenytoin, an anticonvulsant, exhibits nonlinear pharmacokinetics with large interindividual differences. These interindividual differences in dose response can partially be explained by known genetic polymorphisms in the metabolic enzyme CYP2C9 and part of this variability might be accounted for by variable uptake of phenytoin, which is a substrate of P-gp, encoded by the human MDR1 gene.[137] MDR1 polymorphisms C3435T and G2677T are associated with altered P-gp expression in the human placenta and may have clinical consequences owing to genetically determined, variable drug exposure of the fetus.[138] Similarly, MDR1 polymorphism, via altered P-gp expression in BBB, can affect the intracellular concentrations of potentially neurotoxic substances, leading to increased susceptibility for development of Parkinson′s disease.[33] MDR1 polymorphism is not an important genetic risk factor for RA susceptibility, but this polymorphism may have an influence on the activity of the disease and its response to therapy with disease-modifying antirheumatic drugs.[139] Conclusion In conclusion, P-gp is an important component of BBB and placenta barrier, and functions as a protective biological barrier by extruding toxins, drugs, and xenobiotics out of the cell. It not only causes multidrug resistance in cancer but it has also been found to be responsible for MDR of many other clinically important drugs. Altered P-gp expression can lead to increased susceptibility for development of certain diseases also, such as Parkinson′s disease, Alzheimer′s disease, and refractory epilepsy. Hence, targeted inhibition of P-gp may represent an important strategy by which this serious clinical problem can be overcome. It is increasingly being recognized to play an important role in processes of absorption, distribution, metabolism, and excretion of many clinically important drugs in humans. Because of its importance in pharmacokinetics, P-gp transport screening has to be incorporated into the drug discovery process. Inhibition or induction of P-gp by various drugs or herbs can lead to significant drug-drug or drug-herb interactions, thereby changing the systemic or target tissue exposure of the drug. On the other hand, the magnitude of P-gp-mediated drug interactions may be underestimated because of overlapping substrate specificity between CYP-3A4 and P-gp and because of similarities between P-gp and CYP3A4 inhibitors and inducers. But it is clear that drug interaction mediated by P-gp may have a great impact on drug disposition particularly regarding brain and placenta. Thus, care evaluation is desired while indicating mechanism of drug interactions. Genetic polymorphism of P-gp has also been recorded recently, which may affect drug disposition and produce variable drug effects. As drug interactions and genetic polymorphism are important factors taken into consideration for new drug development. Thus, with such a pharmacological relevance associated with P-gp, it may have an impact on drug development in future.References
Copyright 2006 - Indian Journal of Pharmacology The following images related to this document are available:Photo images[ph06003f3.jpg] [ph06003f1.jpg] [ph06003f2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}