 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
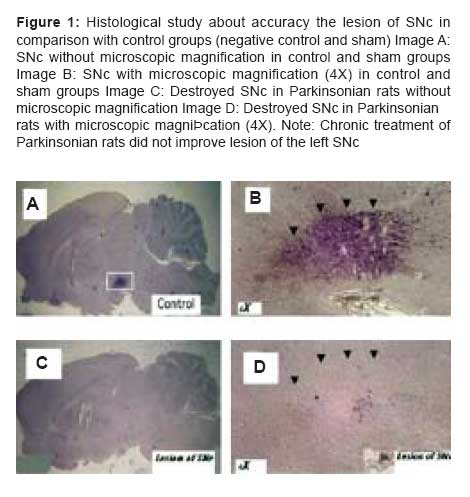
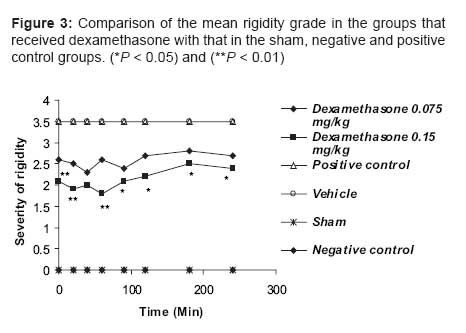
Indian Journal of Pharmacology, Vol. 39, No. 5, September-October, 2007, pp. 235-239 Research Paper Effects of dexamethasone and betamethasone as COX-2 gene expression inhibitors on rigidity in a rat model of Parkinson's disease Mehdi Shafiee Ardestani , Hassan Mehrab1 , Nourallah Sadeghzadeh Department of Medicinal Chemistry and Radiopharmacy, Faculty of Pharmacy, Tehran University of Medical Sciences, Tehran Correspondence Address:Department of Medicinal Chemistry and Radiopharmacy, Faculty of Pharmacy, Tehran University of Medical Sciences, Tehran Date of Submission: 11-May-2007 Code Number: ph07059 Abstract Parkinson's disease (PD) is a neurodegenerative disease in the nigrostriatal pathway of animals and humans and is responsible for most of the movement disorders, including the rigidity. Increasing evidence suggests that an inflammatory reaction accompanies the pathological processes caused by cyclooxygenase-2 (COX-2) seen in many neurodegenerative disorders, including PD. In this study oral betamethasone and dexamethasone were administrated to parkinsonian rats chronically and their effect on rigidity was evaluated. As the results of this study show, both the molecules were able to decrease rigidity. Keywords: Betamethasone, dexamethasone, inflammation, Parkinson's disease Cyclooxygenase (COX) is the first enzyme in the prostaglandin-prostacyclin-thromboxane pathway. It converts arachidonic acid to prostaglandins and thromboxanes, which are collectively known as its metabolites. [1] Three COX isoforms-COX-1, COX-2 and COX-3-have been identified. COX-1 is the constitutive form of COX and performs a housekeeping function to synthesize prostaglandins, which are involved in regulating normal cellular activities. In contrast, COX-2 is the inducible form of COX, as its expression can be induced by inflammatory stimuli or mutagens, tumor necrosis factor-alpha (TNF-α) and the transcription factor CCAAT enhancer binding protein (c/EBP) beta. The brain possesses both COX-1 and COX-2 isoforms. There is COX-2 upregulation during stressful conditions such as cerebral ischemia; it is also upregulated by neuronal apoptosis and neurobehavioral defects. [2] In addition, steroidal anti-inflammatory drugs such as dexamethasone can inhibit COX-2 gene expression. The glucocorticoids have widespread effects because they influence the function of most cells in the body. Glucocorticoids dramatically reduce the manifestations of inflammation. This is due to their profound effects on the concentration, distribution and function of peripheral leukocytes and their suppressive effects on the inflammatory cytokines, such as TNF-α or interleukin-6 (IL-6) and chemokines or other lipid and glucolipid mediators of inflammation. In addition to these effects, glucocorticoids influence the inflammatory response by reducing the prostaglandin synthesis that results from activation of phospholipase A 2 . [3] COX-2 appears to be expressed in dendrites and cell bodies of neurons in several areas of the brain, including the nigrostriatal pathway, CA-1 hippocampus and amygdala nucleus. [4] Among the COX isoenzymes, only COX-2 corresponds to inflammatory and degenerative brain disease. [5] Parkinson′s disease (PD) is a degenerative neurodopaminergic disease in the nigrostriatal pathway of humans. The loss of nerve terminals, accompanied by dopamine deficiency, in this pathway are responsible for most of the movement disorders. [6] Increasing evidence suggests that an inflammatory reaction accompanies the pathological processes seen in many neurodegenerative disorders, including PD. [7],[8] Glial activation is part of a defense mechanism to remove debris and pathogens and promote tissue repair. However, inflammatory activation of microglial cells may contribute to the neurodegenerative process through structural invasion and the release of proinflammatory cytokines, reactive oxygen species (ROS), nitric oxide (NO) and excitatory amino acids at synapses and cell bodies. [9] In cell culture and animal models, inflammation contributes to neuronal damage and nonsteroidal anti-inflammatory drugs (NSAIDs) have been shown to provide some neuroprotection in different paradigms, [10] including PD models. [11] Reactive microglia inhibits neuronal cell respiration via NO and causes neuronal cell death in vitro and in vivo . [12] Investigators have reported an uncertain relationship between COX-2 and its inhibition by NSAIDS and PD. They suggest that chronic use of NSAIDs can decrease the risk of PD. [13] They also suggest that there is a role for COX-2 in degenerative diseases such as PD. [14] In addition, there is the evidence that steroidal compounds like dexamethasone can protect dopaminergic neurons against the lesion. [15] These studies have not examined the effect of COX-2 gene expression inhibitors, such as the steroidal anti-inflammatory drugs dexamethasone and betamethasone, on the rigidity of PD. In the present study, we have investigated the effect of the COX-2 gene expression inhibitors on the rigidity seen in PD. Materials and Methods Animals Drugs and solvents Surgery Stainless steel electrode was then placed in the left SNc and it was destroyed by the electrical lesion maker (Siemens, Germany), using an electrical current (1 mA, 8 s). A lateral lesion of the SNc in each rat caused PD and its associated disorders, such as rigidity, due to the decrease in the inhibitory dopaminergic effects on the caudate nucleus and putamen, which are the main rigidity-inducing neurotransmitter releasing areas in the striatum. [3] Because of these changes in the brains of the lesioned rats, there was rigidity of the limbs on both sides. Following surgery, they were kept in individual cages for 7-10 days and allowed to recover. Experimental procedure It should be pointed out that when an animal had full rigidity (PD) it was given a total score of 3.5. Scores under 3.5 by Murprogo′s Method indicate recovery from rigidity and the effectiveness of the treatment. After the Murprogo′s test, each animal was decapitated and the brain was removed and kept in a 10% formalin solution. Randomly selected brains were cut on a cryostat; 50 µm thick coronal sections were obtained, mounted on glass slides and stained with H & E. Sections were examined under a light microscope to find out the accuracy of lesion of the left SNc. If the lesion was shown not to be in the SNc, any collected data on that particular animal were discarded. Results Statistical analysis Effect of betamethasone on rigidity in parkinsonian rats Effect of dexamethasone on rigidity in parkinsonian rats Comparison of the effect of the two COX-2 gene expression inhibitors on rigidity in parkinsonian rats The test group that received dexamethasone 0.075 mg/kg showed significantly greater decrease in rigidity than the test group that received betamethasone 0.12 mg/kg, with P < 0.01 at 0, 20, 40, 90 and 240 min and P < 0.05 at 90 and 180 min; at 120 min the two test groups showed the same degree of decrease in rigidity. In addition, the test group that received dexamethasone 0.15 mg/kg showed significantly ( P < 0.01) greater decrease in rigidity than the group that received betamethasone 0.24 mg/kg at all the evaluation time points; at 90 and 180 min the difference was statistically significant, with P < 0.05. Finally, we saw significant differences between the groups that received dexamethasone 0.15 mg/kg and that which received betamethasone 0.12 mg/kg, with P < 0.001 for the differences at 0, 20, 40 and 60 min and P < 0.01 at the other time points. Discussion Our observations show that the chronic use of COX-2 gene expression inhibitors, such as steroidal anti-inflammatory agents, caused an improvement in the rigidity of PD in the animal (rat) model. Reduction in rigidity in Parkinsonian rats was more with dexamethasone than with betamethasone and both the drugs were effective in reducing rigidity at all the evaluation time points during the study. Our findings also suggest that there is an important role for COX-2 gene expression in the management of the rigidity of PD. In agreement with our work, a previous study, [18] using postmortem analysis, has shown that COX-2 and prostaglandin E 2 level are increased in the brains of persons who suffer from PD. In another study, Riechman and Hokin [19] suggested that COX-2 causes an increase in the level of acetylcholine in the brain through the production of prostaglandin E 2 and by an increase in the expression of cholinergic markers, such as choline acetyltransferase and vesicular acetylcholine transporter protein. It is worth mentioning that prostaglandins, especially prostaglandin E2, have modulatory effects on adrenergic, noradrenergic and glutaminergic transmission and prostaglandin synthesis inhibitors increase the blood pressure by increasing catecholamine release; for example, using large doses of glucocorticoids in humans may cause insomnia, euphoria and increase in intracranial pressure. [3] In addition, some investigations have shown that COX-2 inhibitors impaired spatial memory by reducing acetylcholine levels in the brain but COX-1 inhibitors do not have any effect on spatial memory in rats. [20] Free radicals and glutamate cause degeneration in SNc and inhibition of these agents by antioxidants or glutamate antagonists protects neurons from degeneration. [21] Other anti-inflammatory effects of steroidal anti-inflammatory drugs possibly include decreasing the production of free radicals and interference with calcium-mediated intracellular events. [3] Neuronal COX-2 overexpression may kill neurons in a cell-autonomous manner and lead to the pathogenesis of PD; [22] In support of this hypothesis is the fact that COX-2 cell-autonomous toxicity may arise from the formation of ROS generated during COX peroxidase catalysis of prostaglandin G 2 conversion to prostaglandin H 2 . That COX-2 cell-autonomous toxicity may arise from the formation of reactive oxygen species generated during COX peroxidase catalysis of prostaglandin G2 conversion to prostaglandin H2 and also electrons donation to COX, co-substrate such as dopamine oxidized to dopamine-quinone and thus neuronal death is happe. In PD, there is evidence of an increase in the oxidative and inflammatory nigral environment, which includes the presence of (COX)-immunoreactive activated microglial cells in the substantia nigra. Microglial cells can also produce and release pro-inflammatory cytokines, in particular TNF-α and cytotoxic molecules, including ROS and NO. [23] Although such responses are nonspecific to lesion type, after 6-hydroxydopamine intrastriatal infusion there is an acute increase in TNF-α in the striatum. [24] In this study we suggest that one of the possible mechanisms for the reduction in rigidity by administration of dexamethasone and betamethasone may probably be a decrease in microglial activation or/and the level of TNF-α, components of the inflammation pathway and free radicals in the striatum region. This hypothesis needs to be further investigated in future studies. In another report scientists have suggested that aspirin and ibuprofen, as nonselective COX-2 inhibitors, significantly attenuate the decreases in dopamine uptake caused by glutamate and thus protect neurons against glutamate excitotoxicity in vitro . [25] These observations suggest that other possible mechanisms exist by which dexamethasone and betamethasone caused reduction in rigidity in the present study. Inhibition of the enzyme COX-2 with inhibition of synthesis of prostaglandin E 2 , reduction in the level of acetylcholine in the brain and, probably, increase in the release of dopamine from dopaminergic neurons in the brain protects dopaminergic neurons from glutamate toxicity similarly to NSAIDS. In support of this, COX-2 inhibition and interference with cellular calcium-mediated events may be effective in achieving neurotransmitter release and recovery from rigidity. Acknowledgments The authors would like to thank Dr. Amin Gravand and Dr. Mostafa Saffari for their kind collaboration in the preparation of this article. References
Copyright 2007 - Indian Journal of Pharmacology The following images related to this document are available:Photo images[ph07059f1.jpg] [ph07059f3.jpg] [ph07059t1.jpg] [ph07059f2.jpg] [ph07059t2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
![[Figure 2]](/showimage?ph/photo/ph07059f2.jpg){kind=link}
{kind=link}