 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
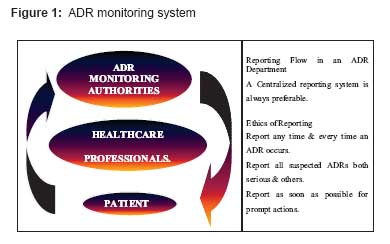
Indian Journal of Pharmacology, Vol. 40, No. 7, Supp. 1, February, 2008, pp. 4-9 Review Article Status of adverse drug reaction monitoring and pharmacovigilance in selected countries Yadav Sachdev Department of Pharmacology, Rajiv Academy for Pharmacy, Mathura, Delhi-Mathura Highway, Chhattikara - 281 006 Code Number: ph08014 The main responsibility of any drug regulatory authority is to ensure the quality, efficacy, and safety of all marketed products. The first two criteria can be established through data obtained from in vitro testing to ensure compliance with acceptable standards and data obtained from animal studies, preclinical and clinical trials involving humans. It is a well-established fact that pre-marketing clinical trials do not have the statistical power to detect rare adverse drug reactions (ADRs) nor do they have significant follow-up to identify delayed ADRs or effects from long-term exposure. In view of this, pharmacovigilance plays a prominent role in establishing the safety profile of marketed drugs. Originally a modest appendix of drug regulation; it has become a major activity now. Definitions "Adverse drug reaction" or an "adverse reaction" means a response to a medicine in the humans or animals, which is noxious and unintended, including lack of efficacy, and which occurs at any dosage and can also result from an overdose, misuse or abuse of a medicine. "Pharmacovigilance": As per World Health Organization (WHO), " Pharmacovigilance is the science and activities relating to the detection, assessment, understanding and prevention of adverse effects or any other drug related problems". Unfortunately, when this term is mentioned, it is very much a case of "Pharmaco what?" There is still a lack of understanding on this topic like how it functions, what are the benefits of sharing ADR knowledge and its purpose and importance. On the other hand, the term "post-marketing surveillance (PMS) study" implies a scientifically rigorous study of a product that is approved for registration in a particular country, designed to produce reliable information about drug safety. It is not appropriate to apply the term to clinical trials of registered products or to studies designed primarily for marketing purposes regardless of the scientific validity of the study design. Post-marketing surveillance studies are generally performed on the initiative of the sponsoring company, but may be suggested or requested by other parties. They should generally be designed to address a specific drug safety question or hypothesis (the latter often identified initially by voluntary reporting). ADR Monitoring Systems: Structural and Functional Aspects The pharmacovigilance concept rests on three pillars:
Four Elements of ADR Reporting Any ADR report should have the following four main elements:
Past Experiences In the past, it was only the developed countries that carried out Research and Development (R&D) activities and new drug launches. The developing countries had a passive "wait and watch" kind of attitude. But now with improving facilities and economics, many of the developed countries are outsourcing their activities, especially those related to clinical trials to these destinations. Hence, now new drugs are almost launched simultaneously here. This changing scenario provides the impetus for more active, vigilant and efficient pharmacovigilance guidelines and control. Pharmacovigilance plays a major role in pharmacotherapeutic decision making, be it individual, regional, national or international. In addition, pharmacovigilance is becoming a scientific discipline in its own right.Issues and Challenges Faced by Developing Countries There is a long list of challenges faced by the developing countries. These are as follows:
Success Factors The success of any pharmacovigilance system depends upon the following factors:
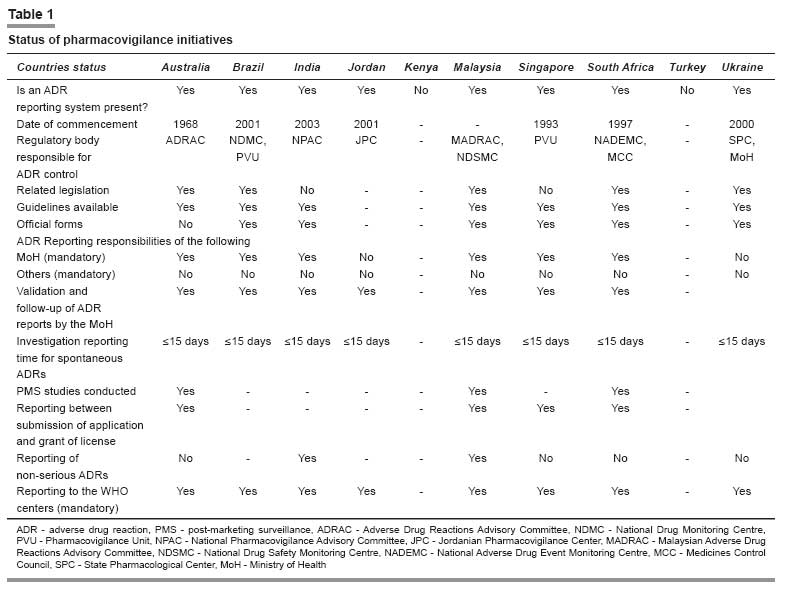
Systems Prevalent in the Selected Countries All the countries except Kenya and Turkey have a pharmacovigilance center or a unit responsible for carrying out the activities related to ADR monitoring, pharmacovigilance and PMS studies. These units are mainly concerned with the collection of spontaneous and suspected ADR reports that have to be submitted on expedite basis. All other reports like non-serious ADRs, etc. need not be submitted on an expedite basis but should be properly archived and made available as and when requested by the regulatory authority. All these reports are then sent to the WHO ADR Monitoring Centre at Uppsala, Sweden, which helps to update the world pool of information on this topic. The status of ADR monitoring and pharmacovigilance in the selected 10 countries can be highlighted through [Table - 1]. ADR Monitoring and Pharmacovigilance Activities in Selected Countries: General Notes Australia The sponsor should report suspected adverse reactions to TGA in an expedited manner in accordance with the following:
The 72-h clock starts from the time of awareness of any personnel of the sponsor. This is considered to have occurred where the sponsor′s review and analysis have been completed and a conclusion is drawn that a significant safety issue exists, or when the sponsor becomes aware of the actions of an overseas regulatory agency. In the period between the submission of a registration application and prior to registration, routine single case expedited reporting is not required except according to the separate guidelines where the product is being used in Australia in a clinical trial. However, in the pre-registration period, information that impacts on the benefit/risk evaluation may become available from the applicant or countries where the medicine is already in use on a compassionate basis, or from countries where the medicine is marketed. Australian Drug Evaluation Committee (ADEC) sponsors are required to submit with their pre-ADEC response a tabulation of any serious unexpected ADRs that are not mentioned in the proposed Australian Product Information and have not been submitted previously. Also cases related to ADRs and pregnancy together with other reports of abnormalities in pregnancy should also be available on request and be included in the next Periodic Safety Update Report (PSUR) together with aggregated data of overall exposure and details of normal/abnormal outcomes. If, in the period between PSURs, a sponsor becomes aware of a signal of a possible teratogenic effect (e.g. a cluster of similar abnormal outcomes), the TGA should be informed immediately. Brazil The main objectives of this unit are:
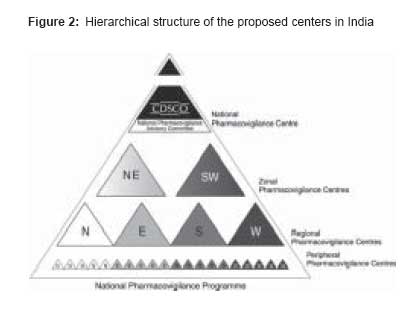
Along with the Department of Inspection of the National Health Surveillance Agency, [2] the PVU monitors the processes of mandatory and voluntary drug recalls in Brazil through the international communication network. Rational drug use has been the object of training and multiple courses sponsored by the National Health Surveillance Agency, with the objective of enhancing health professionals′ capability to prescribe in a rational basis, to identify drug-related problems, and to notify ADRs to the National System of Pharmacovigilance. The PVU made three National Pharmacovigilance Alerts in 2000, 12 in 2001, and 12 in 2002. It translated 90 International Pharmacovigilance Alert versions and published seven Technical Bulletins in 2001 and 10 in 2002. India The Central Drugs Standard Control Organization (CDSCO) [3] is initiating a countrywide pharmacovigilance program under the aegis of DGHS, MoH and Family Welfare, and Government of India. The National Pharmacovigilance Centre at CDSCO shall coordinate the program. The National Centre will operate under the supervision of the NPAC to recommend procedures and guidelines for regulatory interventions. The National Pharmacovigilance Program will have the following milestones:
Periodic Safety Update Reports shall be expected to be submitted every 6 monthly for the first 2 years of marketing in India, and annually for the subsequent 2 years. In addition, training programs and interaction meetings shall be held every 6 months after the initial training [Figure - 2]. All data generated (including reporting forms) will be stored and preserved for the purpose of archiving for a minimum period of 5 years at the ZPCs. The reporting of seemingly insignificant or common adverse reactions would be important because it may highlight a widespread prescribing problem. Jordan The main task is to:
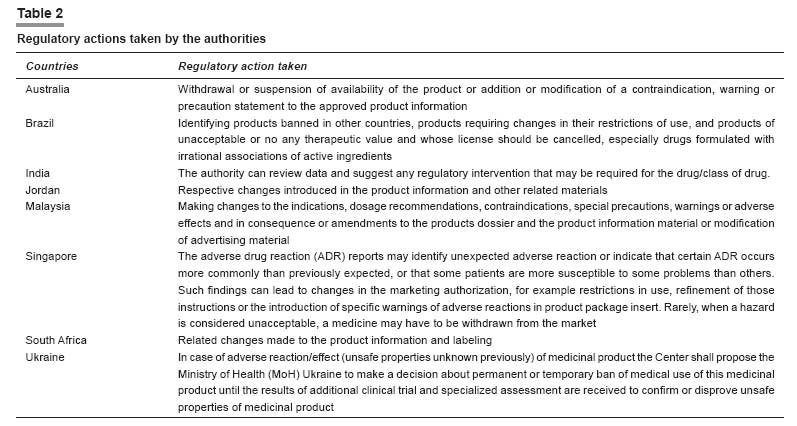
Kenya Malaysia [6],[7] The MADRAC encourages healthcare professionals to report all suspected adverse reactions but it is a compulsory requirement that the marketing authorization holder of a product should inform the DCA of any adverse reactions to the product in accordance to the Malaysian guidelines for ADR monitoring. Wholesalers and Importers Licensed wholesalers and importers in Malaysia should maintain records of every transaction involving a registered pharmaceutical product. The records should be kept for at least 5 years from the date of the transaction. Licensed wholesalers are responsible for keeping records with the following information: (1) date of sale, (2) name and address of the purchaser, (3) name and quantity of the product sold, (4) product registration number, and (5) number of the invoice or delivery order. Likewise, importers in Malaysia are responsible for keeping records with the following: (1) date of importation, (2) name and address of the supplier, (3) name and quantity of the imported product, (4) number of the bill of lading, (5) date of the sale, and (6) name and address of the purchaser. Important: The registration holder is also obliged to submit a "NULL" report at 6-monthly intervals for the first 2 years should there be no ADR reports submitted to them. Registration holders who have registered a product containing a new chemical entity after 1 January 2002 must routinely submit PSURs on that product 6 monthly for the first 2 years after approval in Malaysia and annually for the subsequent 3 years. Singapore The license holder can submit the ADR report to the PVU, using the reporting form prescribed by the unit or the Council for International Organizations of Medical Sciences (CIOMS) I form. The ADR reporting form can be easily downloaded. PSURs may be requested for selected registered medicinal products. PSURs are required to be submitted to the Product Evaluation and Registration Branch (PERB), 6 monthly for the first 2 years after marketing approval, after which they are to be submitted on a yearly basis for the subsequent 3 years. This timeframe may be varied to harmonize periodic safety updates internationally. A CIOMS-I form is an adverse reaction reporting form developed by the Council for International Organizations of Medical Sciences (CIOMS), intended for notifying the regulatory authorities of countries other than the country where the report originated. South Africa After initial receipt of an adverse reaction report, a notice of acknowledgment is sent to the applicant quoting the number assigned to the case report. Any follow-up correspondence from the applicant, relating to the same case report, should be cross-referenced to the assigned database number or to this unique number assigned to the applicant. This is the only reliable way to minimize the duplication of reports submitted by the applicant. Moreover, the applicant should ensure that an internal pharmacovigilance system exists with in the company. Turkey Ukraine Regulatory Action Taken Regulatory authorities can take a wide range of actions in response to a reported ADR. These range from a change in PI to stricter actions like product recalls. Except for Kenya and Turkey, similar actions taken by the regulatory authorities of other countries are tabulated in [Table - 2].Information Dissemination As has been discussed before, an important task of ADR monitoring and pharmacovigilance planning is to collect new information from reliable scientific resources such as marketing authorization holders, healthcare professionals, consumers, international/public bodies, journals, published and updated literature, etc. followed by classifying and analyzing this information and last but not the least circulating its contents as well as any action taken on specific drug to all health sectors. It is this last provision, which provides important impetus to healthcare professionals and general public. The same has been tabulated in [Table - 3]. Recommendations and Suggestions Consolidating ADR monitoring and pharmacovigilance practicesPharmacovigilance is a broad concept, and includes the re-evaluation of marketed drugs, risk management, communicating drug information, promoting rational drug use, and crisis preparedness. It is becoming increasingly important to provide training in all of these activity areas and to carry out intensive monitoring of new drugs to evaluate the risk/benefit.
References
Copyright 2008 - Indian Journal of Pharmacology The following images related to this document are available:Photo images[ph08014f2.jpg] [ph08014f1.jpg] [ph08014t3.jpg] [ph08014t1.jpg] [ph08014t2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}