 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Journal of Pediatric Neurology, Vol. 2, No. 1, 2004, pp. 21-27 Split cord malformation in children undergoing neurosurgical intervetion in India : a descriptive study Raj Kumar 1 , Vinita Singh 2 , Satya Narayan Singh 2
Departments of 1 Neurosurgery, and 2 Neuroanesthesiology,
Sanjay Gandhi Postgraduate Institute of Medical Sciences and
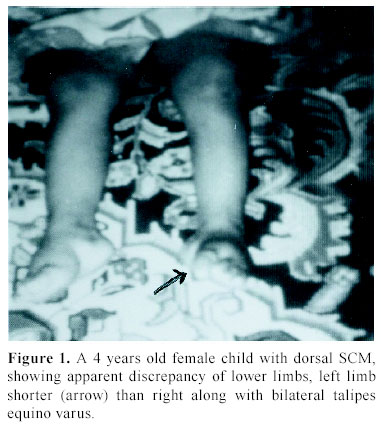
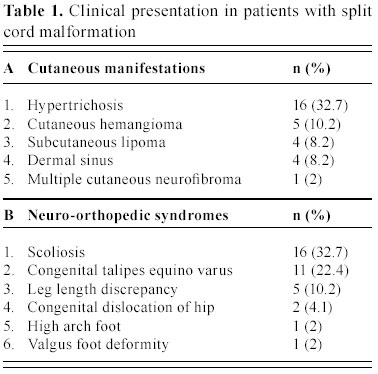
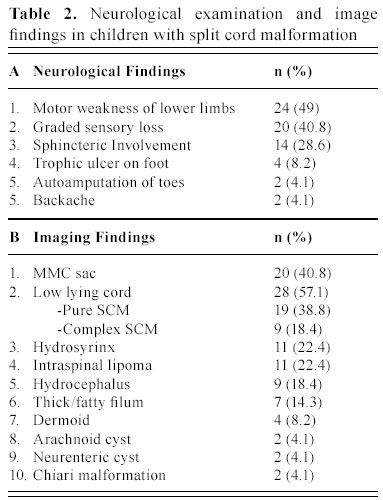
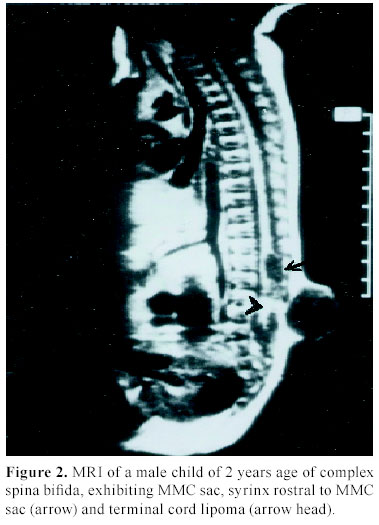
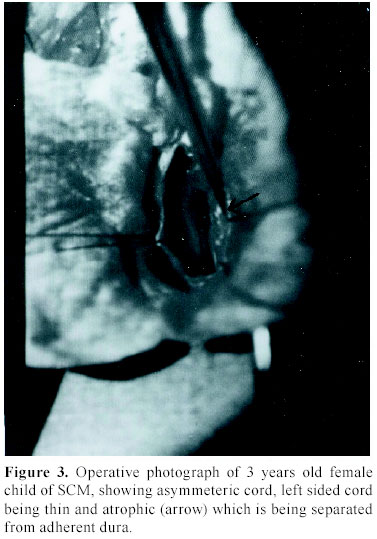
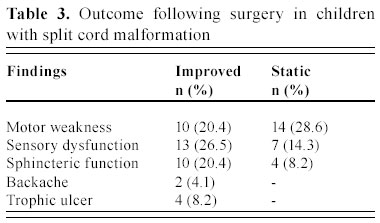
C.S.M. Medical University , Lucknow-226014, India Received: January 31, 2003 . Code Number: pn04005 ABSTRACT To describe the clinical parameters, radiological findings and surgical outcome of 49 cases of split cord malformation (SCM). Forty nine patients of SCM were operated between June 1989 and June 2002, at Sanjay Gandhi postgraduate institute of medical sciences, Lucknow , India out of total 155 cases of spinal dysraphism. Detailed clinical evaluation with magnetic resonance imaging (MRI), preferably craniospinal, was main tool of investigation. All patients underwent excision of bony spur or fibrous septum, repair of associated myelomeningocele sac, when present, excision of associated spinal lesions and detethering of cord. Seven patients needed shunt operation. Patients having minimum 1½ month of follow up were included in this analysis. Mean follow-up period was 3.4 years. Female to male ratio was 1.04:1, with mean age of 4.8 years. The commonest cutaneous manifestation was hypertrichosis in 16 (32.7%) cases and among the neuro-orthopedic syndromes, most frequent were scoliosis in 16 (32.7%) and congenital talipes equino varus in 11 (22.4%) cases. The commonest clinical indicators were lower limb weakness, graded sensory loss, sphincter involvement, and autoamputation of toes and trophic ulcer in 24, 20, 14, two and four patients, respectively. Common imaging findings were low lying cord in 28, neural placode in 19, and SCM with and without meningomyelocele (MMC) sac in 20 and 29 cases, respectively. Common sites of occurrence of SCM were dorsal region in 19 (38.8%) and lumbar in 14 (28.6%) patients. Postoperative complications were cerebrospinal fluid leak, pseudomeningocele, wound infection, and meningitis in 12, eight, four and six patients, respectively. Two patients died in the postoperative period. On average follow up of 3.4 years, motor weakness improved in 10 children and remained static in 14, whereas sensory dysfunction improved in 13 and remained static in seven, and sphincteric function improved in 10 and was static in four cases. Backache dramatically relieved in both children and in all four patients, trophic ulcer completely healed. SCM cases may have association with MMC in significant number of patients hence craniospinal MRI remains the choice, as this will not only delineate complex spina bifida and cord anatomy, but also screen the entire neuraxis for other associated cranial and spinal anomalies. (J Pediatr Neurol 2004; 2(1): 21-27). Key words: split cord malformation, spinal dysraphism, complex spina bifida. INTRODUCTION Split cord malformations (SCM) are spinal cord defects, having double neural tube due to vertical split in the cord (1). SCM is one of the commonest subset of occult spinal dysraphism. Occult spinal dysraphism is defined as skin covered lesion that have no exposed neural tissue and no visible cystic mass (2), barring few cutaneous anomalies, which are indicators of underlying spinal pathology (3). Description of SCM became more enlightening after Pang's classification of SCM. The fundamental error of split cord formation is the formation of accessory neurentric canal between the yolk sac and amnion, which is later invested with mesenchyme to form an endomesenchymal tract that split the notocord and neural plate (4). SCM are of two type, type I consists of two hemicords, each contained within its own dural sheathed tube and separated by a dural-bony median spur, and type II consists of two hemicord housed in a single dura separated by a fibrous median septum (4). Off late, with slight modification of Pang's classification by Kumar et al. (5) it is now obvious that SCM is not merely an entity of occult dysraphism, it may coexist with open spinal dysraphism (complex spina bifida) in significant number of cases. The SCM has variable cutaneous, neurological, orthopedic manifestations and many associated craniospinal anomalies (6). The present study is a further endeavor to document various features, association and variations of SCMs in children. MATERIALS AND METHODS This was a crosssectional descriptive study of 49 operated cases of SCM managed between June 1989 and June 2002 in the Department of Neurosurgery at the Sanjay Gandhi Post Graduate Institute of Medical Sciences, Lucknow , India (SGPGIMS). Total 155 cases of spinal dysraphism, managed in the department of neurosurgery were studied prospectively and retrospectively (143 and 12, respectively) for their outcome. Patient's age ranged from first day of life to 18 years with an average of 4.8 years. Females were almost equal to male patients with ratio of 1.04:1. Four patients with superficial manifestation of meningomyelocele (MMC) were operated outside, where superficial surgery was performed without adequate screening, and they attended our out patient department in view of progressive deterioration in neurological status. All patients underwent detailed clinical examination and investigated preferably with craniospinal magnetic resonance imaging (MRI) to establish the diagnosis. Craniospinal MRI was performed in 30 patients, while 19 cases were referred to our center only with spinal MRI. Cranial computerized tomograpy (CT) scan was performed on suspicion of hydrocephalus in cases with only spinal MRI. There were 20 (40.8%) patients of complex spina bifida (SCM with associated MMC) and 29 (59.2%) cases of pure SCM (SCM without associated MMC). All patients underwent surgery with an aim to excise the bony spur or fibrous septum, repair of MMC sac and detethering of the spinal cord from associated lesions, where present. The cases were evaluated in immediate post operative period and then followed up at regular interval, ranging from 1½ months to 8 years with an average of 3.4 years. The cases with minimum follow-up of 1½ months were included in this analysis. RESULTS There were 25 females and 24 males patients with mean age of 4.8 years. Amongst cutaneous manifestations, hypertrichosis was the commonest seen in 16 (32.7%) of 49 patients of total SCM. Only two children of 20 complex spina bifida cases were showing hairy patch along with MMC sac, whereas it was present in 14/29 children of pure SCM. Other skin markers, exclusively seen in pure SCM, were cutaneous hemangioma in five (10.2%), subcutaneous lipoma in four (8.2%), dermal sinus in four (8.2%) and one patient (2.0%) of SCM had multiple cutaneous neurofibroma. Four patients came with operative scar of previous surgery over the back, who were later detected to be harboring underlying SCM. Amongst neuro-orthopedic syndromes, scoliosis was the most common finding observed in 16 (32.7%) patients followed by congenital talipes equino varus in 11 (22.4%) (Figure 1), leg length discrepancy in five (10.2%) (Figure 1) and congenital dislocation of hip in two (4.1%) cases. High arch foot and foot valgus deformity were noticed in one patient each (2.0%) (Table 1). Neurological examination revealed motor weakness of lower limbs of varying degree in 24 (49%) children and graded sensory loss ranging from 10-100% in 20 (40.8%) patients. Fourteen (28.6%) children showed sphincteric involvement in the form of incontinence of urine and feces. Four (8.2%) patients had trophic ulcer on foot and in two (4.1%) children toes were absent due to autoamputation. Other systemic anomalies noted were bilateral undesended testis, hydronephrosis, and imperforate anus in one case each, though our patients were inadequately investigated for this purpose (Table 2). On imaging, apart from split cord malformation with associated MMC sac in 20 (40.8%) patients (Figure 2), without MMC sac in 29 (59.2%) cases many other coexisting lesions were identified. Low lying cord was the commonest finding seen in 28 (57.1%) patients. Of the 29 (59.2%) patients of pure SCM only ten, who had split cord in dorsal region, had normal position of the conus. In the other nineteen cases conus was lying between L2 and S2, mainly upto lower lumbar level. However, 9/20 (18.4%) patients of SCM with MMC had low-lying cord ending below L2, whereas in 10 (20.4%) patients conus was at the level of L2. Hydrosyrinx was observed in 11 (22.4%) patients of total SCM. Syrinx remained rostral to the defects (Figure 2) and extent was 2-4 vertebral level, except in one child, who had holocord syrinx. No intervention was required for syrinx. Other intraspinal tethering lesions were intraspinal lipoma in 11 (22.4%), dermoid in four (8.2%), arachnoid cyst in two (4.1%), thick/fatty filum in seven (14.3%) and neuroenteric cyst in two (4.1%) patients (Table 2). Nine (18.4%) patients had hydrocephalus, of which 5 (10.2%) were symptomatic with enlarged head and bulging fontanelle, hence, needed CSF diversion before definitive surgery of dysraphism. Two children became symptomatic in postoperative period to manifest with raised intracranial pressure, progression of hydrocephalus was confirmed on serial CT scans and required ventriculoperitoneal shunting. Rest two children remained asymptomatic throughout the follow up period of 4 and 6 years, respectively. Out of 30 children with craniospinal MRI, Chiari malformation and corpus callosum dysgenesis were observed in only two children each, while four children had aqueductal stenosis. Operative procedure and findings In all twenty patients with MMC, excision of thin skin sac or thickened membrane was done. Neural placode was found in 15 (30.6%) children. While detethering the cord and neural placode from dorsally adherent dura, placode or abnormal dysgenetic neural tissue was excised and if possible placode was refashioned into a tubular form. SCM was classified as type I and type II depending on the presence of bony or fibrous septum respectively on radiological or operative findings. Forty (81.6%) patients had type I SCM, while rest nine (18.4%) children had type II SCM. Out of 20 (40.8%) patients of complex spina bifida, 16 (32.7%) had type I SCM and four (8.2%) had type II SCM. Of 29 (59.2%) patients of pure SCM, type I was encountered in 24 (49%) and type II was noted in five (10.2%) cases. In type I variety of SCM, bony spur was extradural, arising from vertebral body to fuse with neural arch and completely excluded from CSF. After laminectomy, bony spur was identified and complete excision was done with small rongeur or micro drill. Both dural tubes were opened in their midline, proximally and distally till single tube could be encountered, usually there were fibrous adhesions with hemicords and dura mainly on medial side. After opening the dura thorough adhesiolysis was performed and dural closure was achieved in such a way so as to make a single dural tube for both the hemicords. In type II SCM, fibrous septum is usually intradural; two hemicords lie in single dural tube. Following laminectomy and durotomy, fibrous septum was excised and then dural closure was done (7). One patient had intramedullary abscess with frank pus and necrosed hemicord while in one child of SCM type I, bony spur was encircling the one half of the cord, along with cord lipoma at the same level and one segment below. In five patients nerve roots were atrophic and in eight cases nerve roots were badly adherent to dura along with arachnoid band causing cord tethering. In three patients one half of the cord was grossly atrophic (Figure 3). Typically, in one patient, neurenteric cyst and cerebellar tissue were found at the level of splitting in the dorsal spine, which were confirmed on histopathological examination. Level of split cord malformation Postoperative complications Surgical outcome DISCUSSION SCM is a rare congenital anomaly that constitute one third of the cases of spinal dysraphism (6), similar incidence was found in our series. The incidence of complex spina bifida (SCM with MMC) was reported to be 78% by Emery and Lendon (8). Surprisingly, in our series it was not such a high rate and observed only in 40.8% cases. SCM is predominantly seen in females (9-12). In our series , female predominance was not marked and the incidence was almost equal (female: male=1.04:1), which may be on account of referral bias to our institute from peripheral hospitals and medical centers. Skin manifestations are very common in case of SCM. Hypertrichosis was the most frequent cutaneous marker seen in 32.7% of patients in this study, as has been reported in many series (9-13). However, 41% of SCM patients had MMC sac as a superficial manifestation. Therefore, it should be kept in mind that the patients with overt spina bifida may harbor underlying SCM in significant proportion of cases and that should be given equal importance in overall management of spinal dysraphism patients. Other skin lesions noticed in patients with SCM include capillary hemangioma, nevus, skin dimple, and subcutaneous mass (9-12). As the incidence of skin marker is very high, the presence of which should alert the physician of a possibility of underlying occult spina bifida, particularly SCM. As Pang (6) has suggested that skin lesions such as hypertrichosis, capillary hemangioma, and dermal sinus tract represent minor aberration in the development of surface ectoderm that is due to the adverse influence of a dorsal endomesenchymal tract, but these aberrations may be overshadowed by chaotic changes in the ectoderm in case of an associated meylomeningocele. Scoliosis is common association with SCM. The incidence of scoliosis varies from 15-75% as reported in different series (10,11,14,15). In our cases the scoliosis was noted in 16 (32.7%) and congenital talipes equinovarus was found in 11 (22.4%). The leg length discrepancy was observed in five (10%) patients. James and Lassman (16) and Guthkelch (14) discussed the orthopedic and neurological syndromes in children with diastematomyelia. Asymmetrical paresis of lower limb is common in patients with SCM (11,14). In our series also 58% had asymmetrical lower limb weakness, whereas only 42% of children manifested as symmetrical weakness of lower limbs . Sphincter dysfunction has been variously reported in cases of SCM (10,15). Bladder and bowel dysfunction had been present in 28.6% of patients in our series and it was based on clinical assessment only. Back pain is the supposed to be the main complaint of the adult patients (13,17). In our series only two adolescent boys presented with backache. It has been seen that neurological deterioration was more marked in patients with SCM who did not undergo early surgery. This deterioration was thought to have been caused by tethering of the cord, secondary to the continued growth of the spinal canal in respect to spinal cord (11). However, the basic pathophysiology responsible for neurological deterioration remains, an intermittent, but chronic, repetitive ischemia to progressive ischemia resulting into spinal cord dysfunction in tethered cord syndrome (18). We also observed that older children had more neurological deficits compared to younger ones. Four children in our study who underwent surgery for meningocele at birth without adequate investigation were having progressive neurological manifestation. Later, they were diagnosed to have underlying SCM, and after resurgery for SCM, their deficits became stabilized. Therefore, we may infer from this data that risk of having neurological deficits increases with age. In asymptomatic adults, symptom and sign may arise after trauma and physical exercise (13,19). Ultrasonography has been used to diagnose SCM in the prenatal period (20). CT myelography and MR imaging are needed for delineating the type of SCM and for showing the associated lesions. CT myelography is superior to MR imaging in defining the type of SCM (15,21). MRI has the added advantage of being noninvasive and very useful in defining the precise anatomy of the cord, nerve roots and dural sac. Because of invasive nature of CT myelography and difficulty in doing the procedure in small children, we opted for MRI study in all our patients. At least one associated tethering lesion unrelated to the SCM is usually seen in majority of these patients, therefore the entire craniospinal axis must be included in screening MR image (6). In one of our patient who had composite SCM, MR imaging did not exhibit the second type II SCM at the dorsal region whereas type I SCM along with MMC was clearly visible 3-4 segment below. In present series, the majority of septa were confined at the dorsal region 19/49 (38.8%). However, many other series have reported frequent occurrence of SCM at lumbar region (9-11,21). Cervical and sacral regions are extremely uncommon. We had only three cases with cervical SCM. The conus medullaries is usually located caudal to L2 (6). Thirty nine (80%) of our patients had their conus ending at or below L2 vertebral level. According to Pang (6), the split segment was greater in patients diagnosed after reaching one year of age than in patients diagnosed before reaching 1 year of age. The mean split length is also greater in type I than in type II SCM. Pang (6) has hypothesized that the neural tube is firmly transfixed by the rigid derivative of the type I endomesenchymal tract and that its subsequent ascent results in a long cleavage in the spinal cord. However, we did not analyzed our cases in that respect, because surgically significant difference was not observed the two types. Eighty percent of patients had at least one associated lesion, responsible for other cause of cord tethering (22). The reported spinal lesions in association with SCM are MMC, meningocele, lipomeningomyelocele, limited dorsal myeloschisis, teratoma, neurentric cyst, lipoma, dermal sinus tract, dermoid, epidermoid, arteriovenous malformation, epidural venous angioma, arachnoid cyst and thick filum terminale (6,10,11,15,21,22). A type I SCM was more frequently associated with a myelomeningocele than a type II SCM and MMC is usually situated below the bony septum (15). In present study, we had similar findings; out of 20 cases of complex spina bifida type I was present in 16 (32.7%) cases and type II in four (8.2%) patients only. Though, in most of the patients (14 of 20) SCM and MMC was at the same level but in six patients SCM was 2-3 vertebral level above the site of MMC. A neurological deterioration is rarely seen after surgery (9,12) and use of microsurgical technique and high speed drill are very helpful in avoiding neural injury. Though the pertinent data is not available for CSF leak from the operative site, but it remains one of the most distressing problem in the postoperative period. We had seen CSF fistula in 12 (24.5%) of patients and pseudomeningocele in eight (16.3%) of cases. Nine of them required re-exploration and duroplasty and rest were managed conservatively. Deaths are generally unrelated to the surgery of the SCM. In present series there were two deaths, one patient died because of fulminant meningitis following CSF fistula in postoperative period and cause of death in the other child was respiratory failure who had complex cervical spina bifida along with associated Chiari malformation. The goal of surgical intervention should be to detether cord and prevent neurological and orthopedic deterioration in the long run. The majority of the patients with SCM will be either stable or better following surgery as was noticed in our series also, however some will continue to worse in these cases screening should be performed to find out another lesion as a cause of progressive deterioration (10,11,22). CONCLUSION SCM cases may have association with MMC in significant number of patients hence CT myelography is better in diagnosing the type of SCM, however craniospinal MRI remains the choice, as this will not only delineate cord anatomy, but also screen the entire neuraxis for other associated cranial and spinal anomalies. In order to avoid irreversible damage to the neural tissue, surgical intervention must be instituted as soon as the diagnosis is made. However, the outcome is less satisfactory in patients with complex spina bifida. ACKNOWLEDGEMENT The authors are extremely grateful to Mr. A.P. Dhar Dwivedi for preparation of this manuscript. REFERENCES 1. Russell NA, Benoit BG, Joaquin AJ, al Fayez N. Adult diastematomyelia. Can J Neurol Sci 1994; 21: 72-74. 2. Korsvik HE, Keller MS. Sonography of occult dysrapism in neonates and infants with MR imaging correlation. Radiographics 1992; 12: 297-306. 3. Reigel DH, Rotenstien D. Spina bifida. In: Pediatric Neurosurgery of the American Association of Neurological Surgeon (ed). Pediatric Neurosurgery (3 rd ed). Philadelphia: WB Saunders, 1994, pp 51-76. 4. Pang D, Dias MS, Ahab-Barmada M. Split cord malformation: Part I: A unified theory of embryogenesis for double spinal cord malformation. Neurosurgery 1992; 31: 451-480. 5. Kumar R, Bansal KK, Chhabra DK. Occurrence of split cord malformation in meningomyelocele: complex spina bifida. Pediatr Neurosurg 2002; 36: 119-127. 6. Pang D. Split cord malformation: Part II: Clinical syndrome. Neurosurgery 1992; 31: 481-500. 7. Kumar R, Singh SN. Spinal dysraphism–trend in northern India. Pediatr Neurosurg 2003; 38: 133-145. 8. Emery JL, Lendon RG. The local cord lesion in neurospinal dysraphism (meningomyelocele). J Pathol 1973; 110: 83-96. 9. Goldberg C, Fenelon G, Blake NS, Dowling F, Regan BF. Diastematomyelia: a critical review of the natural history and treatment. Spine 1984; 9: 367-372. 10. Gower DJ, Del Curling OD, Kelly DL Jr, Alexander E Jr. Diastematomyelia-a 40 year experience. Pediatr Neurosci 1988; 14: 90-96. 11. Miller A, Guille JT, Bowen JR. Evaluation and treatment of disstematomyelia. J Bone Joint Surg Am 1993; 75: 1308-1317. 12. Pang D. Split cord malformations. In: Pang D (ed). Disorders of the Pediatric Spine. New York: Raven Press, 1995, pp 203-252. 13. Russell NA, Benoit BG, Joaquin AJ. Diastematomyelia in adults. A review. Pediatr Neurosurg 1990/91; 16: 252-257. 14. Guthkelch AN. Diastematomyelia. In: Wilkins RH, Rengachary SS (eds). Neurosurgery. New York: McGraw-Hill, 1985, pp 2058-2061. 15. Özek MM, Pamir MN, Özer AF, Kele GE, Erzen C. Correlation between computed tomography and magnetic resonance imaging in diastematomyelia. Eur J Radiol 1991; 13: 209- 214. 16. James CC, Lassman LP. Diastematomyelia. Arch Dis Child 1958; 33: 536-539. 17. Pang D, Wilberger JE Jr. Tethered cord syndrome in adults. J Neurosurg 1982; 57: 32-47. 18. Yamada S, Schreider S, Ashwel S, et al. Pathophysiologic mechanisms in the tethered spinal cord syndrome. In: Holtzman RNN, Stein BM (eds). The Tethered Spinal Cord. New York: Thieme-Stratton, 1985, pp 29-40. 19. Gaskill SJ, Kagen-Hallett K, Marlin AE. Diastematomyelia associated with ectopic renal tissue. Pediatr Neurosci 1988; 14: 108-111. 20. Glasier CM, Chadduck WM, Burrows PE. Diagnosis of diastematomyelia with high-resolution spinal ultrasound. Child's Nerv Syst 1986; 2: 255-257. 21. Harwood-Nash DC, McHugh K. Diastematomyelia in 172 children: the impact of modern neuroradiology. Pediatr Neurosurg 1990/91; 16: 247-251. 22. Guthkelch AN, Hoffmann GT. Tethered spinal cord in association with diastematomyelia. Surg Neurol 1981; 15: 352-354. Copyright 2004 - the Society of Pediatric Science, Yüzüncü Yil University, Faculty of Medicine, Department of Pediatric Neurology, Van, Turkey. Free, full-text articles also available from: http://www.jpneurology.org/ The following images related to this document are available:Photo images[pn04005t2.jpg] [pn04005t1.jpg] [pn04005t3.jpg] [pn04005f3.jpg] [pn04005f2.jpg] [pn04005f1.jpg] |
| |||||||||
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}