 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
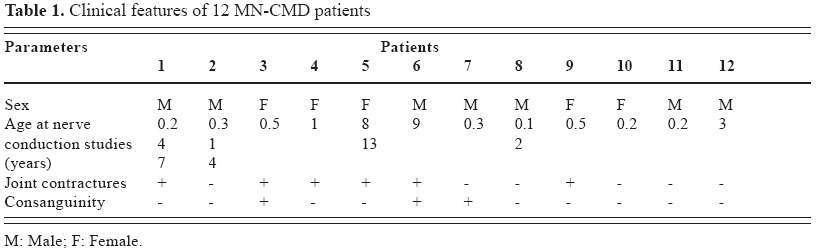
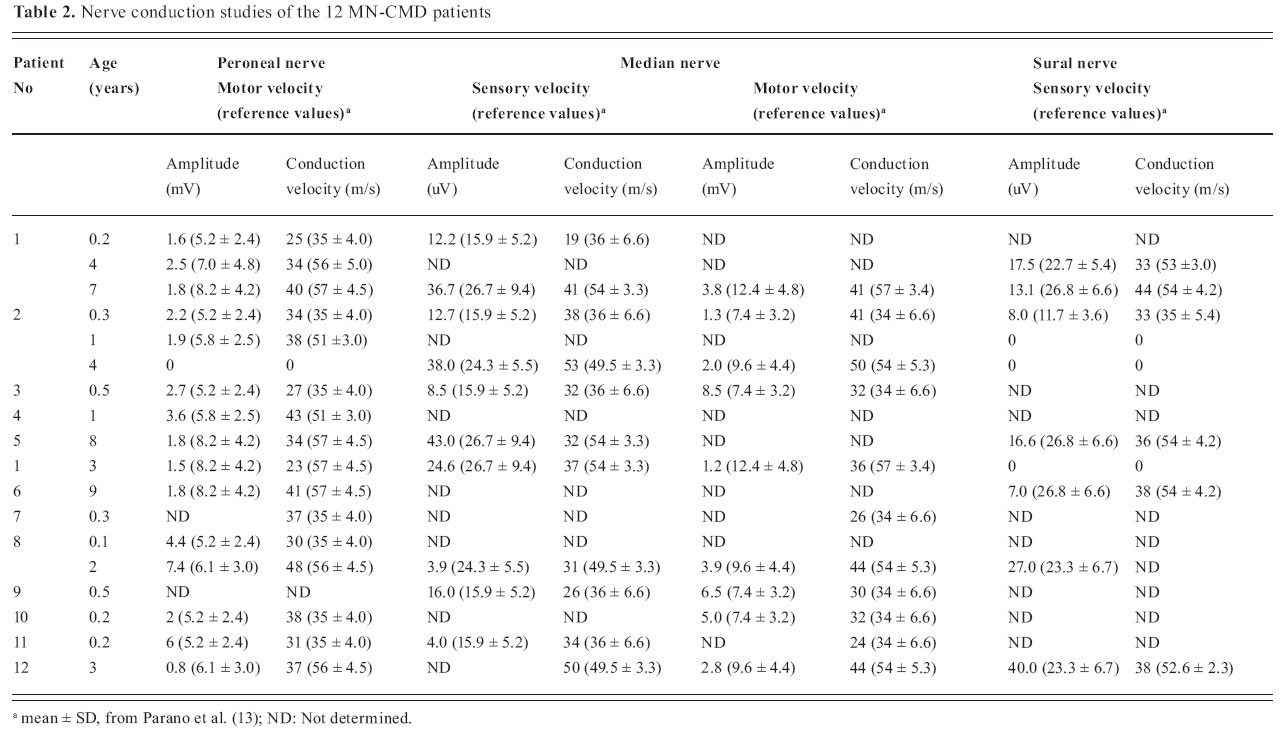
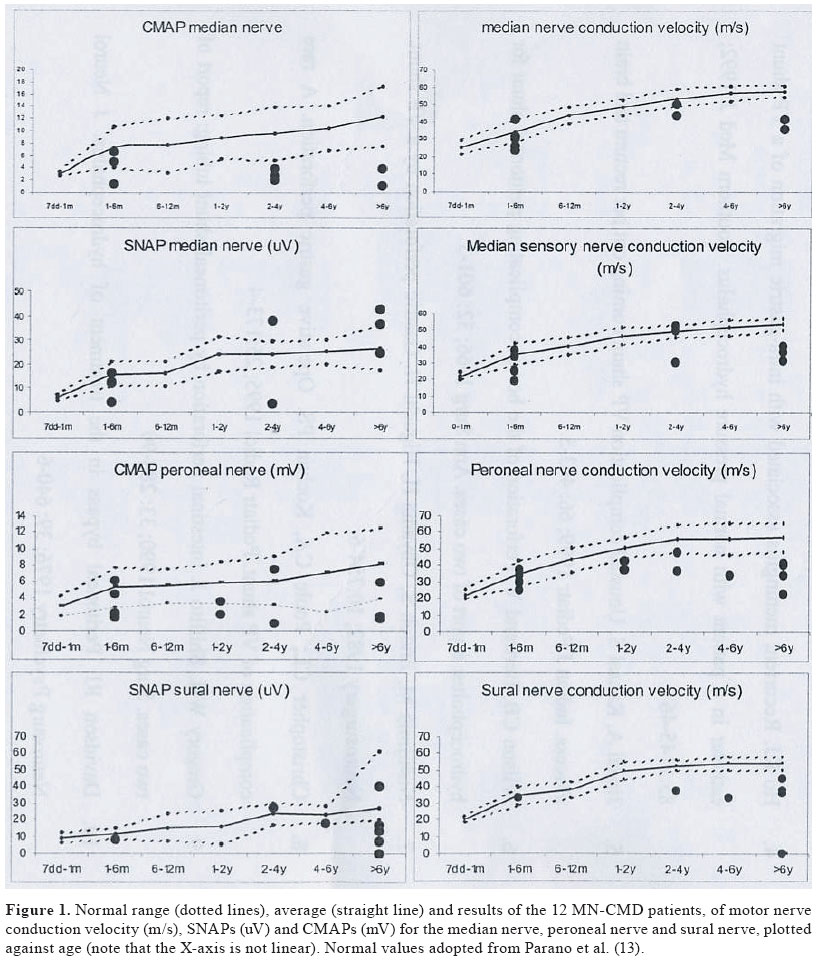
Journal of Pediatric Neurology, Vol. 2, No. 4, Oct-Dec, 2004, pp. 213-218 ORIGINAL ARTICLE Peripheral neuropathy in merosin-negative congenital muscular dystrophy H. Jacobus Gilhuis 1, Oebele F. Brouwer 2, Aad Verrips 3, Machiel J. Zwarts 1 1 Department of Neurology and Clinical Neurophysiology, Neuromuscular
Centre Nijmegen, University
Medical Centre St Radboud, HB Nijmegen, The Netherlands Received: March 22, 2004. Code Number: pn04041 ABSTRACT Peripheral neuropathy in patients with merosin-negative congenital muscular dystrophy (MN-CMD) has been sporadically investigated and has been considered to be motor and demyelinating in nature on the basis of nerve conduction studies. We performed neurophysiologic studies in 12 children with MN-CMD to establish the spectrum and evolution of peripheral nervous system involvement. In our patients, nerve conduction studies for both motor and sensory nerves were near normal in the children younger than six months and abnormal in the older children. The older children had the relatively slowest nerve conduction velocities suggesting a progressive, age-related dysmyelinating neuropathy. We hypothesize that the findings are due to a myelination arrest as a result of insufficient synthesis and maintenance of the peripheral myelin sheath. (J Pediatr Neurol 2004; 2(4): 213-218). Key words: merosin-negative congenital muscular dystrophy, peripheral nervous system. INTRODUCTION The congenital muscular dystrophies (CMDs) form a heterogeneous group of autosomal recessive disorders clinically characterized by generalized muscle weakness with an onset in early infancy, often with congenital contractures. Muscle biopsy shows a dystrophic pattern (1). CMDs without severe mental retardation or malformations of the brain are named “pure” or “classical forms” of CMD (1). Merosin is a heterotrimeric glycoprotein consisting of a heavy chain (laminin a2) and two light chains (laminin b1 and g1). The discovery of a deficiency in the laminin a2 chain of merosin, an extracellular matrix protein, separated classical CMD into merosin-positive and merosin-negative congenital muscular dystrophy (MN-CMD) (2). MN-CMD is caused by mutations in the LAMA2 gene located on chromosome 6q22-23, encoding for the laminin a2 chain of merosin (3). Laminin 2 is expressed in skeletal and cardiac muscles, pancreas, lungs, spleen, kidneys, adrenal glands, skin, testes, peripheral nerves and brain (4-6). In peripheral nerves, merosin is expressed in the basal lamina of Schwann cells whereas its receptors are expressed in the basement membrane of Schwann cells around myelinated axons (4). Clinical manifestations of MN-CMD occur at birth, and consist of severe muscle weakness, markedly delayed motor milestones (more severe than in merosin-positive CMD), and early contractures, often associated with joint deformities. Blood tests reveal elevated serum creatine kinase (2,3,7,8). Peripheral neuropathy has been reported in most, but not all patients with MN-CMD and has been considered to be motor and demyelinating in nature on the basis of nerve conduction studies (9,10). In this study we performed neurophysiologic studies in 12 children with MN-CMD to establish the spectrum and evolution of peripheral nervous system involvement. MATERIALS AND METHODS From 1993 till 2003, 12 MN-CMD patients were identified in four participating hospitals in the Netherlands. The methodology for preparation of histologic sections and immunostaining is described elsewhere (11). All children were found to be negative for merosin in quadriceps muscle on immunostaining (mouse monoclonal antibodies clone MER 3/22 B2 or clone 5H2 on muscle biopsy). All patients showed severe muscle weakness, were wheelchair bound (if older than 1 year), and had an estimated IQ of between 70 and 90. Children older than 6 months had cerebral white matter abnormalities. Nerve conduction studies were repeated once in two children and twice in two other children. Informed consent was obtained for the repeated measurements (Tables 1 and 2). During nerve conduction studies, skin temperature was maintained at a minimum of 30 ºC. Some of the data of six of the children were used in a previous study (12). RESULTS We performed 18 neurophysiologic studies in 12 children to establish the spectrum and evolution of MN-CMD associated peripheral nervous system involvement. Sensory and motor nerve conduction velocities and amplitudes of the peroneal nerve (only motor nerve conduction), median nerve, and sural nerve were compared with normal values obtained from the literature (Table 2) (13). Nerve conduction velocities were normal in the children younger than six months, and abnormal in the older children. The gradual increase in nerve conduction velocities in our patients lagged behind the age-dependent increase of conduction velocities seen in healthy children. The amplitudes of the motor studies were low for both peroneal and median nerve without a clear age-related change. The median sensory amplitudes were normal regardless of patient age. The sural sensory amplitudes were (near) normal when compared to normal confidence intervals or could not be measured (Figure 1). The oldest children had the relatively slowest nerve conduction velocities. Repeated nerve conduction studies in four individual children over the years showed a similar pattern (Table 2). There were no sensory deficits in the three oldest children, in the remaining younger children, no sensory data could be obtained because of young age and low IQ. DISCUSSION We performed nerve conduction studies on 12 MN-CMD children and found nerve conduction velocities were near normal in the children younger than six months and abnormal in the older children. The older children had the relatively slowest nerve conduction velocities as compared to healthy children. The gap in nerve conduction velocities between patients and healthy children increased with time. Repeated nerve conduction studies in individual children over the years showed a similar pattern. The amplitudes in the motor nerve studies differed widely in patients and did not show a clear age-related decrease. This decrease in amplitudes is probably due to secondary axonal injury and loss of muscle tissue due to the myopathy. Sensory nerve studies (excluding the influence of the myopathy) of the median nerve showed conduction velocities slowing with age, but no decrease of amplitudes. This strongly suggests that the neuropathy is due to a myelination problem, and not to axonal degeneration. In case of the sural nerve studies, all amplitudes were near normal or could not be measured, probably due to secondary axonal injury. Shorer et al. (10) found mildly to significantly reduced patients, which they thought was compatible with a demyelinating motor neuropathy. In a nerve conduction study of 2 siblings with MN-CMD (aged 5 and 17 months), the amplitudes were reduced in both children with near normal conduction velocities (14). Mercuri et al. (9) reported a slowing in nerve conduction velocity in a sequential neurophysiological study in one patient, and suggested a failure of the physiological maturation process of myelination of the peripheral nervous system. Di Muzio et al. (15) found reduction of large myelinated fibers, short internodes, enlarged nodes, excessive variability of myelin thickness with no evidence of segmental demyelination in a nerve biopsy of a patient with MN-CMD and slow motor and sensory nerve conductions. In another case of a 19-year-old girl with only mildly reduced laminin 2 chain in muscle but virtually absent in peripheral nerve, nerve conductions were impaired. Sural nerve biopsy fibers showed a ‘globular’ hypermyelination which the authors attributed to alteration of the feedback control during the process of nerve fiber myelination (16). In humans, maturation of myelination of the peripheral nervous system begins during the fourth month of fetal life and it is completed at around five years of age (17). These changes are reflected in the normal increase of conduction velocities during the first five years (Figure 1). In peripheral nerves, Schwann cells cover axons. Merosin is present in the basement membrane of Schwann cells around myelinated axons, and is undetectable in unmyelinated fibers, suggesting that laminin 2 may be important for fiber myelination (18). Merosin is supposed to promote Schwann cell migration and neurite outgrowth, and upregulation of laminin 2 has been demonstrated during peripheral nerve development (19-21). In the peripheral nervous system, Schwann cells normally deposit a basal lamina consisting of laminin among other things. The development of a basal lamina correlates with the Schwann cells’ ability to myelinate axons. For the synthesis and the maintenance of myelin sheath integrity, the expression of laminin a2 receptors like integrins and dystroglycan and the cell-surface bound enzyme lipoprotein lipase (LPL) are essential (17-23). Expression of active cell-surface LPL depends on sufficient extracellular matrix development in order to anchor the LPL molecules effectively (24). Merosin deficiency, resulting in an abnormal basal lamina construction and in a deficient extracellular matrix composition, may thus lead to an arrest of myelin formation of both central and peripheral nervous system. The inability of Schwann cells to myelinate axons sufficiently may be due to the loss of LPL activity, which is necessary to maintain myelin sheath integrity, and for the myelin synthesis, as LPL only works in an intact basal membrane (23,24). Animal studies done so far are not consistent with findings in humans. In the dy/dy dystrophic mouse, an animal model for congenital muscular dystrophy, laminin 2 is deficient in peripheral nerves. Their peripheral nervous system is characterized by “naked” axons in the nerve roots and multiple discontinuities in the basal lamina (25-28). However, another study suggested that the basal lamina is not an absolute requirement for myelination and that laminins a4 and 5 may play a critcal role in myelination instead of laminin a2 (29). A study of laminin chains in rats suggested that laminin a2 plays an important role in postnatal nerve development and axonal regeneration after injury (30). MN-CMD has also been reported in 2 cats. One of the cats which was examined in detail had decreased motor nerve conduction velocities. Biopsy of the nerves showed generalized Schwann cell abnormalities and various stages of demyelination (31). In addition, deficiencies of ligands that interact with laminin a2 such as dystroglycans and integrins also play a role in myelination of peripheral nerves (32,33). It was postulated that b1 integrin participates in the deposition of the immature basal lamina in premyelinating Schwann cells, followed by the expression of -dystroglycan, b4 and b1 integrin during maturation of the basal lamina in myelinating Schwann cells (22). Shorer et al. (10) compared peripheral nerve function between merosin-positive CMD and MN-CMD children and found that all merosin-positive CMD had normal results, whereas 8 of 10 MN-CMD children had reduced nerve conduction velocities. Analysis of the two cases of MN-CMD with normal nerve conduction showed that they produced merosin in reduced amounts, in contrast to the others who had no traces of merosin in their biopsies (10). This confirms that neuropathy is a feature of MN-CMD and may help to differentiate between MN-CMD and other muscular dystrophies. None of our patients achieved ambulation. MN-CMD patients have a more severe muscle weakness when compared with merosin-positive patients. It is unclear if the peripheral nerve involvement contributes to the severity of the clinical condition, as the patients already suffer from a severe myopathy. There were no sensory deficits in the three eldest children, in the other children no gross sensory deficits were found. In summary, we found a pattern of relative age-dependent nerve conduction velocitiy slowing in MN-CMD. This feature, combined with recent findings in biopsies, suggests a disruption in the physiological maturation process of the myelination of both motor and sensory nerves. ACKNOWLEDGEMENTS We thank Dr. R. Ten Houten and Dr K.P.J. Braun for their cooperation, and Dr. J. Pomp and Dr. H.J. ter Laak for their comments. REFERENCES
Copyright 2004 - the Society of Pediatric Science, Yüzüncü Yil University, Faculty of Medicine, Department of Pediatric Neurology, Van, Turkey The following images related to this document are available:Photo images[pn04041t2.jpg] [pn04041t1.jpg] [pn04041f1.jpg] |
| |||||||||
{kind=link}
{kind=link}
{kind=link}