 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Tropical Journal of Pharmaceutical Research, Vol. 3, No. 1, June 2004, pp. 265-277 Research Article Compounded laxative formulations for substituting phenolphthalein with sennosides A & B in solid dosage forms Quintin Verloop1, Wilna Liebenberg1, Andries F. Marais1, Antonie P. Lötter1 and Melgardt M. de Villiers2Φ 1School of Pharmacy, North-West University, Potchefstroom Campus, Potchefstroom 2520, South Africa and 2School of Pharmacy, University of Louisiana at Monroe, Monroe, LA 71209, USA. Code Number: pr04002
Abstract
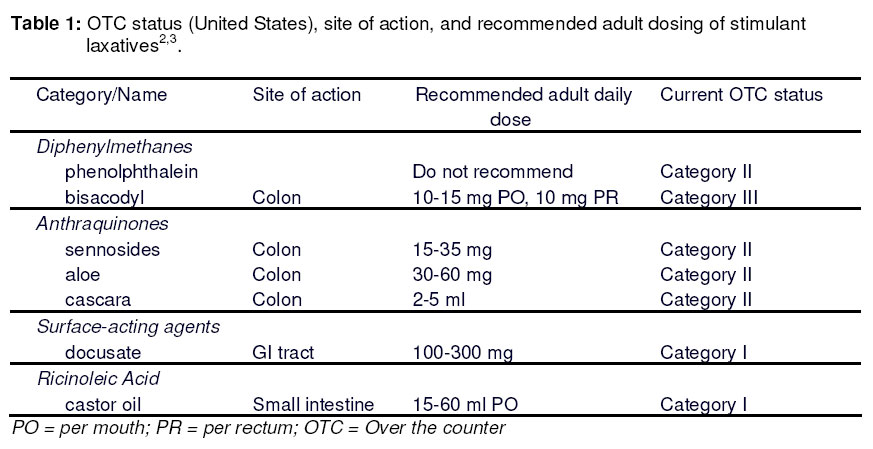
Purpose: Following the discovery of the carcinogenicity of phenolphthalein and the subsequent ban of this compound in several countries this study was undertaken to develop compounded formulations of laxative products containing the stimulant laxatives sennosides A and B. Key words: Compounding; Sennosides A & B; Phenolphthalein replacement; Drug-Excipient Compatibility. Introduction Until recently, the laxative most frequently used because of its fast and efficient effect was phenolphthalein1. Phenolphthalein is a coal tar derivative synthesised in the 1870s and initially used as an acid-base indicator. In 1900, Hungarian pharmacologist Zoltan Vamossy was entrusted with evaluating the toxicity of this compound and he discovered that phenolphthalein was a laxative1. Laxatives are classified as bulk, osmotic, stool softening, lubricant and stimulant laxatives. Phenolphthalein is classified as a stimulant laxative2,3. The stimulant laxatives increase the propulsive peristaltic activity of the intestine by local irritation of the mucosa or by a more selective action on the intramural nerve plexus of intestinal smooth muscle; thus increasing motility4. Since its introduction as a cheap laxative, phenolphthalein has always been seen as a safe drug because only a few cases have been reported where excessive ingestation of phenolphthalein caused adverse effects including an urticarial rash due to ‘toxic epidermal necrolysis’(TEN), erythema multiforme, Stevens Johnson syndrome and abdominal pain5,6 . However, recently phenolphthalein has proven to be a concern, starting in the USA where the National Cancer Institute of America nominated phenolphthalein for study because of its widespread use and lack of adequate testing for carcinogenety. The National Cancer Institute of the USA performed a study on F344/N rats and B6C3F1 mice, giving them 98-99 % pure phenolphthalein with their diet7,8. The rats and mice were studied after 14 days, 13 weeks and 2 years. The 14-day and 13-week studies did not show any difference in the exposed groups of rats and mice, from those of the controls. Under the conditions of the 2-year feed studies there was clear evidence of carcinogenic activity of phenolphthalein in the male F344/N rats based on increased incidences of benign pheochromocytoma of the adrenal medulla and of the renal tubule adenomas. Some evidence of carcinogenic activity of phenolphthalein was found in the female F344/N rats because of the increased incidences of benign pheochromocytomas of the adrenal medulla. There was clear evidence of carcinogenic activity of phenolphthalein in the male B6C3F1 mice based on increased incidences of histiocytic sarcomas and of malignant lymphomas of thymic origin. In the female B6C3F1 mice there was clear evidence of carcinogenic activity of phenolphthalein based on increased incidences of histolytic sarcomas, malignant lymphomas of all types, lymphomas of thymic origin and benign sexcords stromal tumours of the ovary7,8 . In another study it was discovered that there was a significant reduction in fertility in Swiss CD-1 mice, with a decrease in the number of litters per fertile pair and at terminal necroscopy male kidneys were enlarged and right epididymis and testis weights were significantly lower than controls. In males the incidence of abnormal sperm was also increased9. When the findings of these studies were made available the Food and Drug Administration in the USA on 29 January 1999 issued a final rule on the use of phenolphthalein as an OTC laxative proposing a ban of all OTC laxatives containing phenolphthalein10,11. They claim it to be unsafe and misbranded and to be classified as an ingredient (Table 1) which cannot be regarded as safe and effective. This meant that manufacturers of phenolphthalein products then had to seek for a substitute for this popular laxative that now proved to be unsafe. The decision of the FDA has spilled over to other countries such as South Africa where phenolphthalein was banned on 22 February 200212. Prior to the ban as many as 17 products in South Africa contained phenolphthalein in various dosage forms such as tablets, capsules, liquids, gels and granules. These products would now have to be reformulated or removed from the market leaving a huge gap in the range of cheap laxatives currently available. To help with the reformulation and replacement process, this study reports the formulation and stability of several soliddosage forms containing sennosides A and B (Senna). This laxative was chosen as an alternative because it falls within the same category of stimulant laxatives (Table 1) as phenolphthalein. Senna is also widely used and still regarded as safe and effective. Materials and Methods
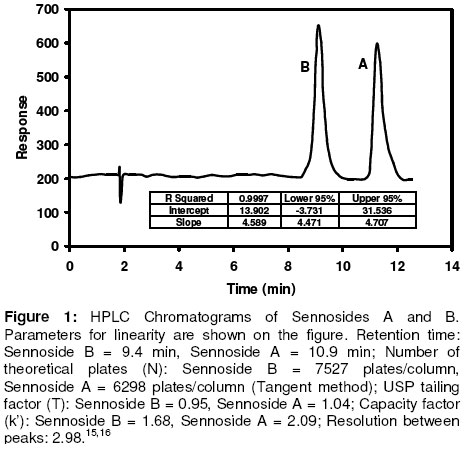
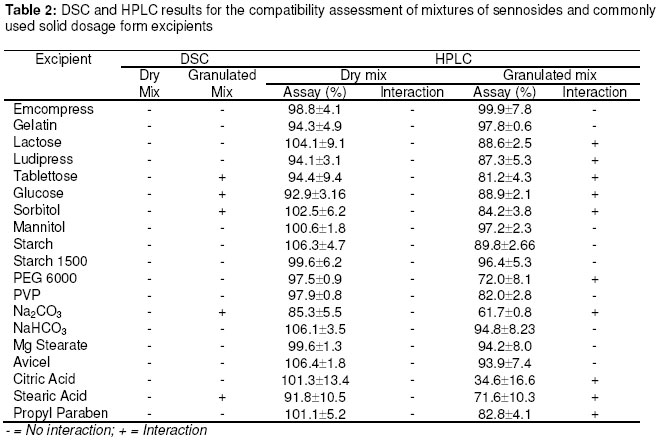
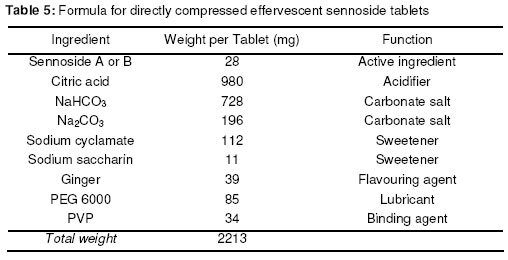
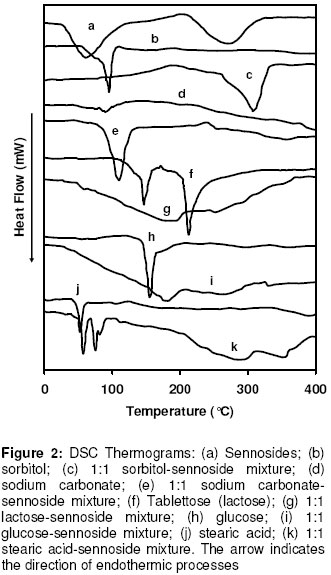
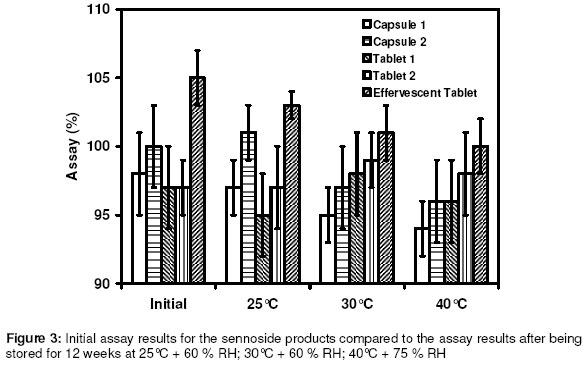
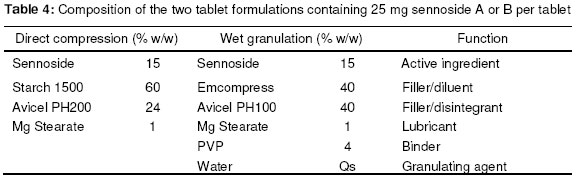
Materials In this study calcium sennosides A and B obtained from Ciba Geigy (Johannesburg, South Africa) were used. Chemically, this compound belongs to the group of glycosidelinked anthraquinones and consists principally of two stereoisomers, namely sennoside A and sennoside B. The following excipients were used: Lactose (Tablettose, Meggle Excipients, Germany and Ludipress, BASF, South Africa), dibasic calcium phosphate dihydrate, (Emcompress, JRS Pharma LP, New York, USA), mannitol, microcrystalline cellulose (Avicel pH 101 & PH200, FMC Corporation, Ireland), pregelatinised starch (Starch 1500, Colorcon, USA), polyvinyl pyrrolidone (Kollidon 25, BASF), gelatin, glucose, polyethylene glycol 6000 (BASF), magnesium stearate, stearic acid, sorbitol, sodium bicarbonate, sodium carbonate, citric acid, and propyl paraben. All other solvents and chemicals used were of analytical grade and were used as received. Drug-Excipient Compatibility Studies Assessment of possible incompatibilities between an active drug substance and different excipients forms an important part of the preformulation stage during the development of a solid dosage form13 . Successful compatibility studies require a good experimental design that furnishes the required information with the minimum of experimental effort. In this study 1:1 w/w samples of active and excipients were intimately mixed in a mortar with a pestle, either as dry powders or powders wetted with a small amount of water. The granulated samples were dried in an oven for 1 hour at 60°C. These mixtures were then evaluated by differential scanning calorimetry (DSC) and high performance liquid chromatography (HPLC). For DSC analysis samples with a mass between 2-4 mg were measured and crimped into aluminium seal pans. The sealed pans were placed in a Shimadzu DSC-50 Differential Scanning Calorimeter (Shimadzu, Japan). The sealed pans were heated at a heating rate of 10 °C/min and under a nitrogen purge with a flow rate of 30 ml/min. DSC thermograms of the 1:1 w/w mixtures were compared to the thermograms of the individual excipients and sennosides. The criteria used to determine if an excipient was incompatible with the sennosides were the following: the peaks have either elongated or broadened, extra peaks occurred, peaks shifted or disappeared, or there was a shift in melting points of endotherms and exotherms as result of thermal behavior. These changes in peaks are due to incompatibility with the sennosides. For HPLC analysis of the sennosides the method described by Muffat et al. was used with a few alterations14. The instrument used was a HP1050 series HPLC equipped with a HP1050 quaternary gradient pump, HP1050 autosampler, HP1050 diode array detector and Chemstation data acquisition and analysis software (Agilent, California, USA). The column used was a Luna C18-2 µm column, 250 x 4.6 mm (Phenomenex, California, USA); mobile phase: methanol: acetonitrile: buffer (240:160:600 v/v/v); buffer: 0.01 M tetra n-butyl ammonium iodide (3.68 g/l) in water, pH adjusted to 7 using NH4OH; flow rate: 1.0 ml/min; retention time: 9.4 min and 10.9 min for the 2 sennoside peaks (Figure 1); injection volume: 10 µl; detection: UV at 270 nm. Formulation of Solid Dosage Forms In this study capsules, tablets and effervescent granules containing sennosides were prepared using those excipients that were compatible with the active ingredient. To prepare capsules the ingredients were weighed and mixed together in a Turbula mixer (WA Bachofen, Switzerland) for 5 min. A hand-operated capsule filling machine was used to fill the mixtures into size 0 capsules. Table 2 shows the formulae used to manufacture the sennoside capsules. One directly compressed and one wet granulated tablet formula, Table 3, were made for stability purposes after several trial and error batches of a number of different formulas were made. For direct compression, the ingredients were weighed and mixed together in the Turbula mixer for 5 min. The mixture was then compressed into tablets at the predetermined mass and hardness. With wet granulation the sennosides, PVP, Avicel and Emcompress were weighed and mixed together in a mortar with a pestle. Distilled water was added and mixed until the blended powders were moistened sufficiently. The wet mass was spread out on drying trays and dried in an oven at 60°C. After drying, the granules were passed through a 2 mm mesh screen, magnesium stearate was added and the mixture was then mixed in a Turbula mixer for 5 min. The granules were then compressed into tablets at the predetermined mass and hardness. To prepare the effervescent tablets only water-soluble excipients were used as shown in Table 5. To prepare the mixture for compression, citric acid, sodium bicarbonate, sodium cyclamate, sodium saccharin, PVP and sennosides were weighed and mixed together with a mortar and pestle. Water was added and the mixture was mixed vigorously while effervescence occurred. The wetted mixture was sieved through a 2 mm mesh and dried at 60 ºC. The flavor, sodium carbonate and PEG 6000 were weighed and added to the wetted mixture after it had been cooled. The powder was mixed thoroughly and again sieved through the 2 mm mesh sieve. The mixture was then compressed into tablets at the predetermined mass and hardness. Stability Testing of Solid Dosage Forms Tablets, capsules and effervescent tablets were stored at 25 °C (60 % Relative Humidity (RH)), 30 °C (60 % RH) and 40 °C (75 % RH). They were tested at 0 (initial), 6 and 12 weeks. The following tests were used to evaluate the tablets: hardness, friability, assay, uniformity (mass, thickness and diameter), loss on drying, disintegration and dissolution. For the capsules the following tests were used: uniformity (mass of capsule and empty capsule mass), assay, disintegration, loss on drying and dissolution. The hardness of 20 tablets of each formula was measured with a hardness tester (Pharma Test, type PTB 103, Switzerland). The friability of the tablets gives a good indication of how they will withstand transport and handling. Twenty tablets were weighed and were put into the Roche friabilator for 4 min after the tablets were dusted and weighed again. The percentage weight loss was then determined. For the assay test standards were made equivalent to the amount of sennosides in the capsule, tablet or effervescent tablet. The samples consisted of powdered tablets or capsules equivalent to the mass of one tablet or capsule. The amount of sennosides in the standard and samples were then determined using HPLC. The USP states that the tablets should have 90 –110% of the labelled amount of sennosides15 . The same limits were used for the capsules or effervescent tablets since there is no mention of sennoside capsules or effervescent tablets in the USP. To determine the weight uniformity twenty tablets and capsules were weighed individually and their mean weight was determined. The thickness and diameter were determined with a Pharma Test, type PTB 103, Switzerland. Not more than two tablets or capsules are permitted to differ from the mean by a percentage greater than stated in British Pharmacopoeia14 . For the capsules, individual capsules were weighed and the contents of the capsule emptied. The capsules were then washed with ethanol and left to dry. The empty capsules were then weighed. The difference in weight showed the weight of the capsules. To measure the loss on drying, about 1 g of the capsule contents and powdered tablets was weighed. The capsule contents or powdered tablets were put in an oven and dried to constant weight at 105 °C. They were allowed to cool and were then weighed again. The percentage weight loss was determined and should not exceed 6 % according to the BP16 . In addition the disintegration time of six tablets and capsules were also measured using the standard method of the USP15 . Method 1 of the USP (basket method) was used to determine the dissolution rate of the solid dosage forms. Before dissolution testing each tablet or capsule was weighed. The dissolution medium was 900 ml water which was maintained at 37 °C. A basket was used to hold the tablet or capsule which rotated at 100 rpm. The samples were obtained with a pipette, followed by replacement with an equal volume of dissolution medium. The samples were analysed using HPLC. The dissolution rate was not measured for the effervescent tablets but the effervescence time was measured by placing the tablet in 250 ml distilled water at ~20ºC and recording the time when no more gas bubbles evolved from the tablet. The pH was also measured after the tablet had completely dissolved. Statistical Evaluation of Data Statistical analysis was performed to determine significant differences at a 95% confidence level (p<0.05) (Microsoft Excel, Microsoft Corporation, USA). Results and Discussion Excipients for the formulation of sennoside tablets, capsules and effervescent tablets were chosen from the Handbook of Pharmaceutical excipients17. Examples were chosen from each of the major excipient categories based on cost, availability, and acceptability. Compatibility between Sennosides and Excipients A summary of DSC results for the evaluation of the compatibility between the sennosides and commonly used solid dosage form excipients are given in Table 2. A (-) indicates no interaction between sennosides and excipient and a (+) indicates that there was an interaction. In the dry form no interactions occurred. In the wet granulated samples however five interactions were discovered because glucose, sodium carbonate, sorbitol, stearic acid and lactose (Ludipress® and Tablettose®) showed incompatibilities. These interactions were derived from the disappearance of the dehydration, 72 °C, and melting, 276 °C, peaks of the sennosides (Figure 2) in the mixtures. The glucose-sennoside mixture in a wet granulated sample showed only one peak at 183°C, indicating the disappearance of the glucose and sennosides peaks. Pure sodium carbonate showed one peak at 87°C (Figure 2) which shifted to 109°C in the wet granulated sample. Sorbitol showed peaks at 96 °C (Figure 1) which moved to 422 °C and 308 °C in the mixture. Stearic acid showed one peak at 56 °C, but an extra peak appeared in the wet granulated sample at 74 °C. Tablettose showed peaks at 146 °C and at 213 °C, typical for lactose monohydrate dehydration and melting respectively. The wet granulated lactose samples showed only one peak at 192 °C. HPLC analysis was used to confirm the incompatibilities obtained by DSC. The chromatogram of the sennosides (Figure 1), showed a peak at 9 min (sennoside B) and a peak at 11 min (sennoside A). Interference by individual excipients with the HPLC of the sennosides was tested by injecting samples of the pure excipients. None of the excipients tested had retention times close to that of the sennosides. The results listed in Table 2 confirm that there was no interaction between the sennosides and excipients in the dry state. However, in the granulated samples statistical analysis showed that propyl paraben, sodium carbonate, stearic acid, citric acid, PEG, lactose, glucose and sorbitol were incompatible with the sennosides. Especially, HPLC showed that the recovery for sodium carbonate (62 %), stearic acid (72 %) and citric acid (35 %) was very low in the wet granulated mixtures. Formulation of Sennoside Capsules Hard gelatin capsule is a convenient, consumable pharmaceutical package that can be used to produce stable, uniform unitdose forms because it is robust and can be filled with a variety of materials to yield stable products. In Table 3 the composition of two capsule formulations containing 15 mg sennoside and filled into size 0 capsules with a capacity of ~ 0.3-0.7 g, depending on the density of the powders, are listed. Due to lower density, larger particle size and higher porosity of microcrystalline cellulose compared to Starch 1500, the capsule fill mass of Formula 1 (± 360 mg) was less than that of Formula 2 (± 575 mg). Using the criteria set by the BP16 , the average weight of capsules weighing more than 250 mg cannot deviate from each other by more than 5 %. The weight deviation of both Capsule 1 and 2 fell within this range at time 0 and up to 12 weeks when stored at the three test conditions (25 °C + 60% RH; 30 °C + 60 % RH; 40 °C + 75% RH). However, there were significant differences (p < 0.05) between the disintegration times of Capsule 1 and 2 initially and after storage; but the disintegration time was still within the 30 min required by the BP and USP. Throughout, the disintegration time of Formula 2 was consistently longer than that of Formula 1. This could be because of the superior disintegration properties of microcrystalline cellulose and the difference in particle size between Avicel, ~100 µm, and Starch 1500, ~ 52 µm17,18. The smaller starch particles filled the capsules more densely forming a much denser plug that took longer to disintegrate. Loss on drying measurements showed that Capsule 1 had an average percentage weight loss of 4-5 % while Capsule 2 had a percentage loss of 8-10 %. This may be because starch is slightly more hygroscopic than microcrystalline cellulose17 . However, the moisture uptake of both formulas was within the limits set by major pharmacopoeia and did not influence the sennoside content of the capsules. The USP15 states that sennoside tablets should not contain less than 90 % and not more than 110 % sennosides. Since there are no limits for sennoside capsules, the limits for the tablets were used to evaluate the content of the capsules. Both Capsule 1 and 2, Figure 3, complied with this specification under all the conditions for which the capsules were tested. Potential problems with the availability of the sennosides from the capsules were tested by measuring the dissolution rate of the drug from the capsules. The limits for the dissolution of sennoside tablets according to the USP state that not less than 75% of the labelled amount should be dissolved after 120 min. These limits were also used for the sennoside capsules. All of the dissolution profiles fell within the limits specified by the USP for sennoside tablets because the dissolution of both formulas were more than 75% of the labelled amount of sennoside dissolved within 60 min. As an example the initial dissolution profiles for capsules 1 and 2 compared to that obtained after storage at 40 °C for 12 weeks are given in Figures 4. The lag time for the dissolution from Capsule 2 (Figure 4) correlated with the differences in disintegration times. Comparison of the dissolution profiles for the two formulas at 25 ºC, 30 ºC and 40 ºC showed that the sennosides seem to be completely dissolved after 15 min from Capsule 1 and within 45 min from Capsule 2. To quantify this difference, the dissolution profiles were compared using a mathematical method to calculate the similarity between two dissolution profiles19 .
The similarity factor (f2) is a logarithmic reciprocal square root transformation of the average of the squared vertical distances between the test (T) and reference (R) mean dissolution values at each dissolution time point, and is a measurement of the similarity in the percent (%) dissolution between the two curves. Values of f2 between 50 and 100 indicate sameness of the two dissolution profiles19 . Comparison of the dissolution profiles of Capsule 1 constantly produced f2 values below 50 which is an indication of dissimilarity between the initial profile of this formula and the profile both at 25ºC and 40ºC after 12 weeks. The f2 values calculated for Capsule 2 at all the test conditions were between 50 and 100 and the dissolution profiles were therefore not significantly different. Formulation of Sennoside Tablets The majority of pharmaceutical dosage forms are marketed as tablets. To the consumer tablets offer the convenience of administration, handling, identification and proper dosage; to the manufacturer relative low-cost and high speed production. In this study one direct compression formula and one wet granulation formula were made for stability purposes after several trial and error batches of a number of different formulas were tested. The formulae for sennoside tablets are shown in Table 4. Tablets from both formulations complied with pharmacopeial standards for content (mean: 96 %; minimum: 93 %; maximum: 105 %), weight uniformity, tablet thickness and diameter, hardness (mean 59 ± 4.1 N), moisture content, and friability. These properties did not change significantly after storage for 12 weeks at elevated temperature and humidity. The disintegration times for the wet granulated formula (20 min) were significantly longer than that of the directly compressed formula (10 min) and for the granulated tablet formula there was also a significant increase (p<0.05) in disintegration time after storage at elevated temperatures.The longest disintegration time (40 min) was measured for tablets stored for 12 weeks at 40 ºC. This may be due to the fact that PVP, used in the formulation, has a high binding strength and when kept at higher temperatures the binding strength increased due to a process known as polymer curing. Polymer curing is a process whereby the reaction between a polymer and a liquid causes the polymer to harden over time18 . The dissolution profiles for the direct compressed formula differed from the wet granulated formula. The direct compressed formula dissolved more rapidly than the wet granulated formula (Figure 5). This correlated with the difference in disintegration times. The USP states that for the dissolution of sennoside tablets, not less than 75% of the labelled amount of sennosides must be dissolved after 120 min and both formulae complied with these limits. There were also no significant differences in dissolution profiles for the direct compressed formula initially and after 6 and 12 weeks. In contrast the dissolution of the wet granulated tablets decreased with time, being the slowest after 12 weeks at 40 ºC, but these dissolution results still complied with the limits stated by the USP. Comparisons with the initial dissolution profile produced f2 values for the direct compressed formula of 50 to 100. This confirmed that the dissolution profiles for this formulation did not change. The f2 values for the wet granulated tablets however, differed all from the initial profile (f2 < 50). These differences correlate well with the differences in disintegration time. The f2 values also decreased gradually and the tablets stored at the highest humidity and highest temperature (40 °C + 75% RH) has the lowest f2 value (29) when compared to the initial dissolution profile. Although these differences were significant, all the dissolution tests complied with the dissolution criteria set by the USP for sennoside tablets. Formulation of Sennoside Effervescent Tablets Effervescence is defined as the evolution of bubbles of gas in a liquid, as the result of a chemical reaction when solids dissolve. Effervescent mixtures have been known and used medicinally for many years. Effervescent tablets are convenient, easy to use, pre-measured dosage forms that are already in solution when ingested. Another significant advantage of effervescent tablets is that they can be used to mask unpleasant flavours and convert solids into liquids for easier consumption. In this study, the formula in Table 5 was used after trying out several batches using trial and error. These trial batches had disintegration times that were too long, or the powder stuck to the punch and die during tableting. The effervescent tablets were stored for 12 weeks at elevated temperatures and relative humidity in aluminium tubes containing silica gel as a desiccant in the HDPE lids and in HDPE containers without silica gel in the lid. Tests for loss on drying, hardness and time of effervescence were done on the tablets stored in these containers, to determine the effect of moisture on the tablets. After 12 weeks the sennoside content of the tablets were not significantly different from the initial assay and all the tablets complied with the USP specification of between 90-110% active per tablet. The pH (5.7) of the solutions formed after effervescence was determined. It also did not change with time. After 12 weeks the tablet weight, tablet thickness and diameter, and time for effervescence to be completed (2 – 3 min) did not change for the tablets stored in containers containing the desiccant. However, the time for effervescence to be completed significantly increased (p<0.05) for the tablets stored in containers without a desiccant. The time for effervescence to be completed increased from an initial value of ~ 2 min to ~ 5 min after 12 weeks at 40 °C + 75% RH. ConclusionThe results of the compatibility study showed that thermal analysis is useful for the determination of reactions between sennosides and excipients, especially when combined with the quantitative analysis of the drug with HPLC. Both DSC and HPLC confirmed no incompatibilities in the dry mixtures. The only inconsistent results between DSC and HPLC were propyl paraben and citric acid that showed an interaction by HPLC and not by DSC. Compatibility evaluation shows that dry powder mixtures for filling capsules and for direct compression of tablets can thus be used to formulate sennosides A & B into tablets. Wet granulation can be used to formulate tablets and capsules containing sennosides, but propyl paraben, sodium carbonate, stearic acid, citric acid, PEG, and sugar derivatives such as lactose, glucose and sorbitol should not be used in these formulations. Using the results from the compatibility study it was possible to compound two sennoside capsules which complied with the criteria set by the USP for sennoside tablets although there were slight differences in the dissolution rate, disintegration times, and stability. Sennoside tablets were also successfully manufactured using both direct compression and wet granulation methods. The directly compressed formula had a faster disintegration time and dissolved more quickly. However, the flow properties of the wet granulated formula were better, which should make it the more preferred method for large scale manufacture. Both formulae were stable for the 12 weeks tested at elevated temperatures up to 40°C + 75% RH and should therefore be viable for compounding sennoside tablets. In addition to capsules and tablets, effervescent tablets that dissolved quickly and remained stable for 12 weeks if stored with desiccant in the containers were also successfully compounded. These tablets would be ideally suited for treating patients that have difficulty with swallowing tablets and capsules. AcknowledgmentThe authors would like to acknowledge the financial assistance from the National Research Foundation of South Africa. References
Copyright @2002-2006. TJPR Faculty of Pharmacy, University of Benin, Benin City, Nigeria The following images related to this document are available:Photo images[pr04002t5.jpg] [pr04002t3.jpg] [pr04002f4.jpg] [pr04002t2.jpg] [pr04002f3.jpg] [pr04002f2.jpg] [pr04002t1.jpg] [pr04002t4.jpg] [pr04002f1.jpg] [pr04002f5.jpg] |
| |||||||||

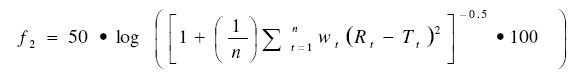
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}