 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
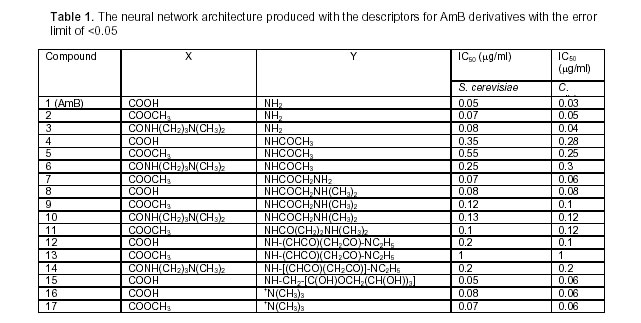
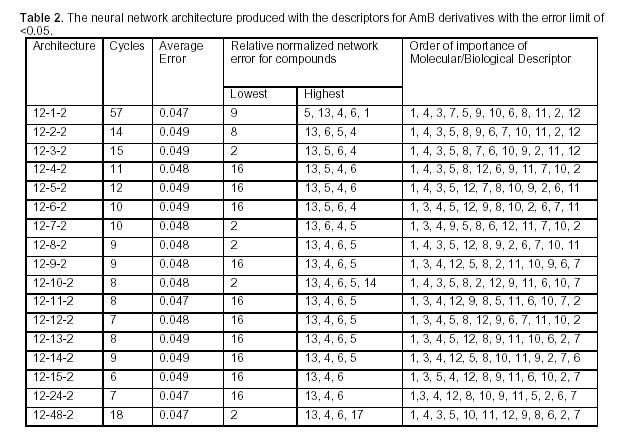
Tropical Journal of Pharmaceutical Research, Vol. 4, No. 2, December 2005, pp. 517-521 Research Article Evaluation of SAR for Amphotericin B Derivatives by Artificial Neural Network S Sardari*1 and M Dezfulian2 1 Dept of Biotechnology, Pasteur Institute, Tehran, Iran 2 Dept of Biology, Ahvaz University, Ahvaz, Iran *Corresponding Author, ssardari@hotmail.com Code Number: pr05014 Abstract Aim: This study was designed to investigate the role of several descriptive structure-activity features in the antifungal drug, amphotericin B and analyze them by artificial neural networks. Key words: Amphotericin B, SAR, Artificial Neural Network. IntroductionAmphotericin B (AmB) is a polyene discovered in 1956 and produced by Streptomyces nodosus. This agent is the first effective compound available for systemic mycosis, used against serious mycotic infections. AmB is regarded by many as the drug of choice for the seriously ill patients, in spite of a number of major drawbacks. It is nephrotoxic, poorly tolerated, and has such low solubility in biological fluids that it has to be formulated as a colloidal suspension in bile salts and administered as a slow infusion (Sarba, 1990). In addition, to reduce the toxicity, lipid-based and liposomal formulations of AmB have been either marketed or are undergoing further studies (Hiemenz, et al., 1996; Wasan, et al., 1997). Nevertheless, AmB is still claimed by many to be the best standard, against which other agents should be compared. It has a broad spectrum of activity and is fungicidal against most species, at least in vitro. The newer azole antifungals have still not replaced it for many indications, particularly in the immunocompromised hosts. The polyenes exert their effect by associating with the eukaryotic membrane sterols and disrupting membrane integrity, as can be shown by a rapid efflux of potassium ions from treated cells (Hunter, 1995). It is believed that AmB inserts into membrane by having a high affinity for ergosterol in the fungal membrane. This affinity is lower for cholesterol, therefore, AmB shows a greater effect on fungi in low concentration than on host cells. A better understanding of the structural features can lead to making derivatives with higher affinity to ergosterol and increased fungitoxicity compared to mammalian cell. Adverse effects would be important in rational design of new compounds with improved profile. A number of substituted amphotericin B molecule have been synthesized and their activity studied by principle component analysis methodology (Cheron, et al., 1988). The QSAR of amphotericin B and its 16 semi-synthetic derivatives (Table 1), obtained by modification at carboxyl and amino groups, were lead to certain assumptions on the role of net molecular charge on the results of biological tests (antibiotic action, erythrocyte K+ release, haemolysis) by numerical methods (Cheron, et al., 1988). (Figure 1) A number of computational methods have been found useful in the area of structure-activity relationship (SAR) and structure-property relationship (SPR) (Hansch, et al., 1995, 2001; Mize, et al, 1995; Estrada, et al., 2001; Langgard, et al., 2000). In this study, we report the application of neural network methodology to correlate SAR data and find new correlations between the biological and physicochemical descriptor values such as parachor, lipophilicity, SMILES, log P and the membrane activity for amphotericin B and its derivatives. The following is the list of such descriptors: 1- S. cerevisiae YK50, the dosage that led to 50% potassium release from yeast cells compared to 100% value for the boiled cell suspension; 2- Log P, logarithm of n-octanol/water partition coefficient; 3- Erythrocyte H50, the dosage that led to 50% haemolysis; 4- Erythrocyte EK50, the dosage that led to 50% potassium release from erythrocytes compared to 100% value for the lysed cells; 5- Positive charge on Y functionality at pH= 7.4; 6- Negative charge on X functionality at pH= 7.4; 7- Positive charge on X functionality at pH= 7.4; 8- Lack of free -COO; 9- Ergosterol 410 UV band position in the presence of the compound tested; 10- Cholesterol 410 UV band position in the presence of the compound tested; 11- SMILES code, Simplified Molecular Input Line Entry System, a chemical notation system based on the principles of molecular graph theory; 12- Parachor, an additive physical property of a substance related 9to its molar volume, determined by the kind, number of atoms and their manner of arrangement. An artificial neural network (ANN) is a biologically inspired algorithm designed to simulate the way in which the human brain processes information. ANN, are composed of many processing elements (PE). Each processing element has inputs, transfer functions and output. Processing elements are connected with coefficients and are arranged in layers, i.e., input layer, output layer and hidden layers in between (Sardari, and Sardari 2002). Application of ANN in pharmaceutical research is a new field with novel potentials to be discovered. A variety of areas have been described to benefit from such algorithm predictions that range from industrial design systems to optimal formulation prediction and SAR evaluations (Rowe, et al., 1998; Agatonovic-Kustrin, et al., 2000). Here, the design and application of ANN for antifungal drug AmB and its synthetic derivatives is reported. A set of 17 structurally related AmB derivatives was selected and their experimentally derived membrane activity on erythrocyte cells and antifungal values against Candida albicans and Sacharomyces cerevisiae were collected (Cheron, et al., 1988). A standard feed-forward network, with back propagation rule and with single hidden layer architecture was chosen applying the EasyNN, 8.01 (1999-2001). The number of neurons was kept minimum to avert an over-fitting problem, which is usually produced by more weights due to higher numbers of neurons in input and hidden layers. However, to produce the optimum architecture, powerful enough to model the functions and not create errors more than 0.05%, the number of hidden layer neurons were varied from 1-48. The following architectures were produced that met the error limit condition using least number of calculation cycles (Table 2). Higher numbers of hidden layer did not improve the performance, yet can decrease the speed of calculation. This finding is in accordance with previous reports (Ripley, et al., 1996). The descriptor parameters, including Log P and parachor, and SMILES coding were calculated using the Toolkit for Estimating the Physicochemical Properties of Organic Compounds, v. 1.0, John Wiley & Sons, Inc. The best architecture, in terms of cycles of calculation was 12-15-2. The importance is relative to the greatest sum of absolute weights connected to the next layer of the architecture. Therefore, importance of an input descriptor is determined by the sum of the absolute values of the weights of all the outgoing architecture connections from the input node to the next layer. Among the most important factors were biological descriptors 1, 3, and 4, that correlated best with the model produced by ANN. Among the chemical and structural descriptors, positive charge on Y substitution was found to be the most important, followed by lack of availability of free carboxyl and parachor. The least important chemical and structural descriptors are 2, 7, and 11. The correlation coefficients between the experimental and the predicted IC50 value pertaining to all the compounds for S. cerevisiae and C. albicans obtained by ANN methodology are 0.94 and 0.82 respectively. This indicates the applicability of this method in prediction studies. Compounds 4, 5, 6, and 13 corresponded to the highest error that was generated during the training cycles. The least error values were attributable to compounds 2 and 16. The results of this study show the ability of ANN algorithm in assigning the biological and physicochemical descriptors to the activity prediction of large antibiotic molecules such as AmB. The findings of this study reveals the significance of various factors participating in the desired antifungal activity; therefore such method can be applied in drug design and development of different derivatives with better profile of activity, less toxicity and more potency. In this regard, the produced architectures using ANN modeling can be applied to in silico study of the biological activity for new derivatives prior to synthetic steps. References
Copyright 2005. Pharmacotherapy Group, Faculty of Pharmacy, University of Benin, Benin City, Nigeria. The following images related to this document are available:Photo images[pr05014t1.jpg] [pr05014t2.jpg] [pr05014f1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}