 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
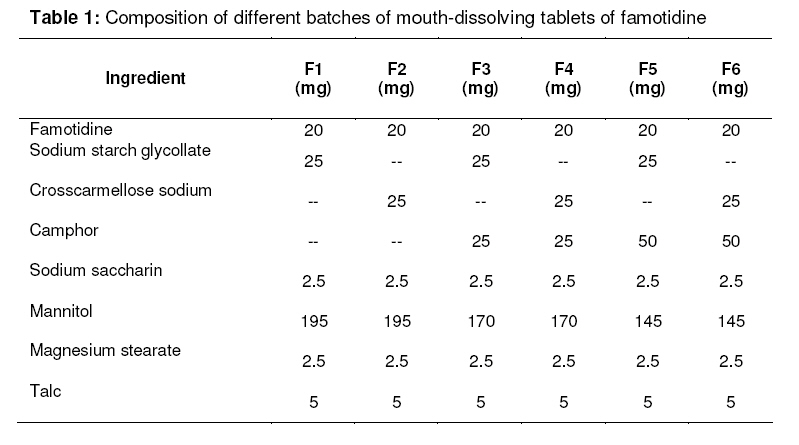
Tropical Journal of Pharmaceutical Research, Vol. 7, No. 4, December, 2008, pp. 1185-1189 Research Article Development and characterization of orodispersible tablets of famotidine containing a subliming agent S Furtado*, R Deveswaran, S Bharath, BV Basavaraj, S Abraham and V Madhavan M.S Ramaiah College of Pharmacy, M.S.R. Nagar, M.S.R.I.T Post, Bangalore-560054.India. Received: 10 April 2008 Revised accepted: 25 August 2008 Code Number: pr08042 Abstract
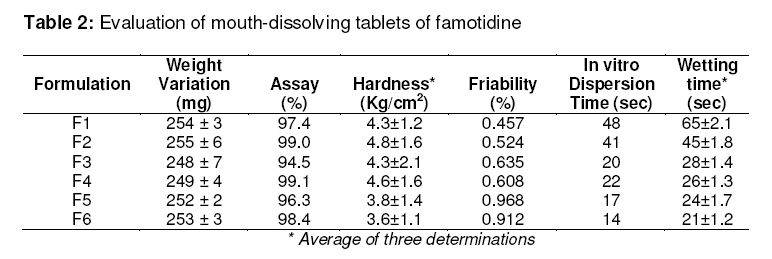
Purpose: The purpose of the present research was to the effect of camphor as a subliming agent on the mouth dissolving property of famotidine tablets. Keywords : Mouth dissolving tablet, Famotidine, Subliming agent, Super disintegrant, Camphor. INTRODUCTION Tablet is the most widely used dosage form because of its convenience in terms of selfadministration, compactness and ease in manufacturing. Patients often experience difficulty in swallowing conventional tablets when water is not available nearby. Furthermore, paediatric and geriatric patients may also encounter inconvenience in swallowing it1 . ‘Mouth dissolving’ (MD) or ‘melt in mouth’ tablets are a perfect fit for these patients as they immediately release the active drug, when placed on the tongue, by rapid disintegration, followed by dissolution of the drug2,3,4. Mouth dissolving tablets combine the advantage of both liquid and conventional tablet formulations allowing the ease of swallowing the drug in the form of liquid dosage form. Some drugs are absorbed from the mouth, pharynx and esophagus as the saliva passes down into the stomach. In such cases, the bioavailability of the drug is significantly increased over those observed in the conventional tablet dosage form. The basic approach to the development of mouth dissolving tablets is the use of superdisintegrants such as crosscarmellose sodium and sodium starch glycolate. Another approach used in developing MD tablets is maximizing the pore structure of the tablet matrix. Freeze drying and vacuum drying techniques have been tried by researchers to maximize the pore structure of the tablet matrix. However, freeze drying is cumbersome and yields a fragile and hygroscopic product. Vacuum drying along with the sublimation of volatilizable ingredient has been employed to increase tablet porosity. While designing dispersible tablets, it is possible to achieve effective taste masking as well as a pleasant feel in the mouth. The main criterion for MD tablets is the ability to disintegrate or dissolve rapidly in saliva of the oral cavity in 15 to 60 seconds and have a pleasant mouth feel5 . Famotidine is a highly selective H2 receptor antagonist with properties of inhibiting gastric acid secretion and healing gastric & duodenal ulcers6 . Since the aqueous solubility of the drug is 0.1%w/v at 20°C7, it gives rise to difficulties in the formulation of dosage forms leading to variable dissolution rates 8, 9. In the present study, an attempt was made to develop mouth dissolving tablets of famotidine and to investigate the effect of subliming agent on the release profile of the drug in the tablets. MATERIALS AND METHODS Materials Famotidine was obtained as a gift from Tonira Pharmaceuticals, Ankhleshwar. sodium starch glycollate (SSG) and crosscarmellose sodium were also gifts from Recon Health Care Ltd, Bangalore. Camphor, sodium saccharin, mannitol, polyvinyl pyrollidone(PVP), talc and magnesium stearate were purchased from S.D. Fine Chemicals, Mumbai, India. Method Formulation of mouth dissolving tablets The orodispersible tablets of famotidine were prepared using the subliming agent, camphor, SSG and crosscarmellose sodium as superdisintegrants, mannitol as a diluent, sodium saccharin as sweetening agent, alcoholic solution of PVP (10%w/v) as binder and magnesium stearate with talc as a flow promoter (see Table 1). The drug and other ingredients were mixed together, and a sufficient quantity of alcoholic solution of PVP (10%w/v) was added and mixed to form a coherent mass. The wet mass was granulated using sieve no. 12 and regranulated after drying through sieve no. 16. Granules of the formulations containing either of the superdisintegrants but without camphor (F1 or F2) were dried in a tray dryer (Tempo instruments and equipments, Mumbai) at 60°C for 30 min. resulting in localized drying. Since the melting point of famotidine is 163-164 °C7 , drying the granules at 60°C does not affect the stability of famotidine. Other granular formulations (F3 to F6) contained a subliming agent and were dried at room temperature, 20-22°C for 8hrs. The final moisture content of the granules was found to be between 4-5%10, 11, which was determined using an IR moisture balance. The dried granules were then blended with talc, magnesium stearate and compressed tablets using a 8mm punch rotary tablet machine (Rimek, RSB-4 mini press Cadmach, Ahmedabad, India). Tablets from formulations F3 to F6 were further vacuum-dried at 60ºC until they reached constant weight. During drying, the camphor sublimed with the formation of a porous structure on the surface of the tablet. Evaluation of the tablets Hardness The crushing strength of the tablets was measured using a Monsanto hardness tester. Three tablets from each formulation batch were tested randomly and the average reading noted. Friability12 Ten tablets were weighed and placed in a Roche friabilator and the equipment was rotated at 25 rpm for 4 min. The tablets were taken out, dedusted and reweighed. The percentage friability of the tablets was measured as per the following formula,
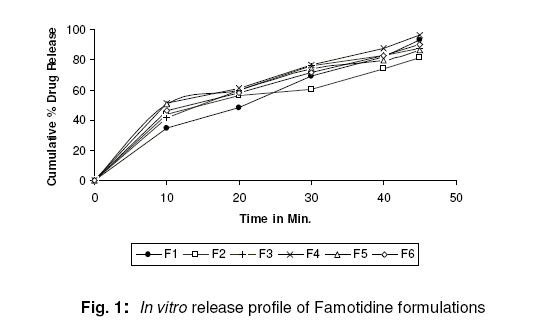
Weight Variation Randomly, twenty tablets were selected after compression but before vacuum-drying (in the case of F3 to F6) and the mean weight was determined. . None of the tablets deviated from the average weight by more than ±7.5% (USP XX). Drug content Twenty tablets were weighed and powdered. An amount of the powder equivalent to 20mg of famotidine was dissolved in 100ml of pH 6.8 phosphate buffer, filtered, diluted suitably and analyzed for drug content at 265nm using UV-Visible spectrophotometer (UV 160-Shimadzu, Japan). Wetting time13 A piece of tissue paper (12cmx10.75cm) folded twice was placed in a Petri dish (Internal Diameter=9cm) containing 9ml of buffer solution simulating saliva pH 6.8, which had the following composition, NaCl (0.126g), KCl (0.964g), KSCN (0.189g), KH2 PO4 (0.655g) and urea (0.200g) in 1 Litre of distilled water14 . A tablet was placed on the paper and the time taken for complete wetting was noted. Three tablets from each formulation were randomly selected and the average wetting time was noted. The results are tabulated in Table 2. In vitro dispersion time15 In vitro dispersion time was measured by dropping a tablet in a 10ml measuring cylinder containing 6ml of buffer solution simulating saliva fluid (pH 6.8). Dissolution studies In vitro drug release studies of all the formulations were carried out using tablet dissolution test apparatus (USP TDT 06 PL, Electrolab, Mumbai) at 50rpm. Phosphate buffer pH6.8 was used as the dissolution media with temperature maintained at 37±1ºC. Samples were withdrawn at different intervals, diluted suitably and analyzed at 265nm for cumulative drug release using Shimadzu UV-Visible spectrophotometer. RESULTS Table 2 shows that all the formulated tablets exhibited low weight variation. Addition of a subliming agent had no pronounced effect on hardness and increased friability of the tablets. The wetting time, in vitro dispersion time of the tablets were also considerably reduced in tablets containing camphor (Table 2). The drug content of all the formulations was found to be between 94.5 -99.1% which was within the acceptable limits as per USP XXVII. The in vitro dissolution profile (Fig. 1) indicated faster and maximum drug release from formulation F4. DISCUSSION Tablets were prepared using superdisintegrants alone (F1 & F2) and in combination of a superdisintegrant and a subliming agent, camphor (F3 to F6). Addition of camphor in the formulation improved the tablet properties with respect to wetting time and in vitro dispersion time. Comparatively increased concentration of camphor in formulation F5 and F6 showed relatively decreased wetting time and in vitro dispersion time which may be attributed to faster uptake of water due to the porous structure formed thus facilitating the disintergrant to bring about faster disintegration. CONCLUSION Overall, the results suggest that suitably formulated mouth-dissolving tablets of famotidine containing camphor as a subliming agent along with a super disintergrant (crosscarmellose sodium or sodium starch glycollate) can be achieved. The tablets exhibited good in vitro dispersion and wetting properties in presence of subliming agent. Thus the present study demonstrated potentials for rapid absorption, improved bioavailability, effective therapy and patient compliance. ACKNOWLEDGEMENT The authors are thankful to Gokula Education Foundation for providing necessary facilities to carry out the research work. REFERENCES
© Pharmacotherapy Group, Faculty of Pharmacy, University of Benin, Benin City, 300001 Nigeria. The following images related to this document are available:Photo images[pr08042f1.jpg] [pr08042t2.jpg] [pr08042t1.jpg] |
| |||||||||


{kind=link}
{kind=link}
{kind=link}