 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Tropical Journal of Pharmaceutical Research, Vol. 8, No. 1, February, 2009, pp. 33-41 Research Article Formulation of chitosan-based ciprofloxacin and diclofenac film for periodontitis therapy Mohammed Gulzar Ahmed*, NM Harish, R Narayana Charyulu, Prabhakar Prabhu Department of Pharmaceutics, NGSM Institute of Pharmaceutical Sciences, Paneer Deralakatte, Mangalore 574160, Karnataka, India Received: 15 July 2008 Revised accepted: 10 October 2008 Code Number: pr09006 Abstract
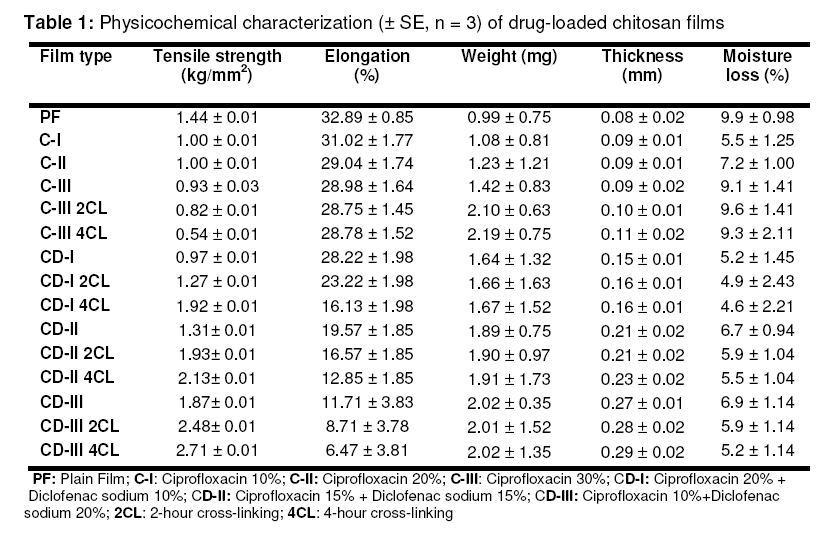
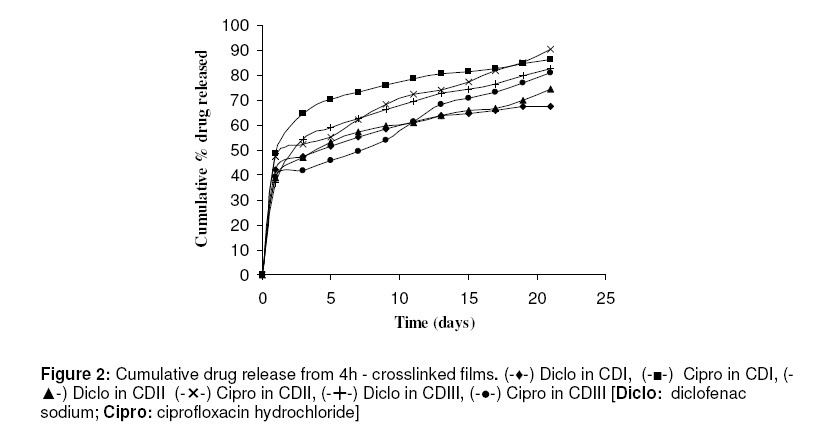
Purpose: This study was designed to develop and evaluate chitosan films containing ciprofloxacin and diclofenac sodium for the topical treatment of periodontitis. Key Words: Chitosan, Ciprofloxacin, Crosslinking, Diclofenac, Films, Topical delivery, Periodontitis. INTRODUCTION Periodontal diseases are infections affecting a significant proportion of people in all populations1 . The presence of periodontal pathogens such as Porphyromonas gingivalis, Prevotella intermedia and Actinobacillus actinomycetemcomitans are responsible for periodontal destruction2 . Therefore, an objective of periodontal treatment is to suppress or eliminate subgingival periodontal pathogens. This is generally achieved through subgingival debridement, resulting in the reduction of the total bacterial load3 . However, some patients may experience continued periodontal attachment loss4 , and this may be due to some periodontal pathogens that are inaccessible during mechanical periodontal therapy. On the other hand, it is conceivable that local and/or systemic administration of effective antimicrobial agents may enhance the outcome of mechanical therapy5 . Systemic antimicrobials such as adjuncts to mechanical therapy have had a positive effect on clinical as well as microbiological parameters6 . But the impact of this approach is reduced by the fact that the antibiotic is normally difficult to maintain in therapeutic concentrations at the site over the course of the treatment period. Moreover, systemic antibiotic therapy carries with it the risk of the host developing resistance. Due to these negative effects, the use of local drug delivery devices containing antibiotics which can maintain therapeutic concentrations at the site of infection is an approach that may be explored. This could enhance the therapy of periodontal diseases while also reducing side effects7 . Pharmacological agents applied locally for the treatment of periodontitis are targeted to several areas such as bacteria in periodontal pockets, soft tissue walls of the pocket, and the exposed root cementum. Experimental evidence suggests that many forms of local delivery are not able to deliver medications to all these locations. For example, agents in mouth rinses and those used for irrigation do not predictably reach beyond 5 mm into the periodontal pocket8 . On the other hand, gaining access to the anatomical boundaries of the pocket does not mean penetration to the target bacteria, because bacterial aggregates (biofilm) impair diffusion or inactivate pharmacological agents. Also, rapid turnover of gingival crevicular fluid in the sulcus rapidly reduces the concentration of the locally placed antimicrobial agent9 . It has been shown that the fluid present in a 5 mm pocket is replaced about 40 times an hour. This high rate of clearance represents a major obstacle to maintenance of effective concentrations of antimicrobial agent within the pocket10 . Earlier, it was hypothesized that local production of prostaglandins and other metabolites of arachidonic acid within the periodontal tissues contributes to alveolar bone resorption in periodontitis. Research findings have shown that inhibitors of prostaglandin production, such as nonsteroidal anti-inflammatory drugs (NSAIDs), could affect the course of bone loss in periodontal disease11 . Data from prospective animal experiments and human studies support this concept, and indicate that NSAIDs can reduce gingival inflammation and reduce alveolar bone resorption12 . There is also evidence that systemic administration of antibiotics and NSAIDs are effective in altering the progression of certain forms of periodontitis12 . Chitosan is a hydrophilic biopolymer obtained by alkaline deacetylation of chitin, a major component of arthropod shells, and possesses favorable properties such as nontoxicity, biocompatibility, bioadhesivity and biodegradability. Moreover, chitosan itself possesses antimicrobial activity13,14 . Ciprofloxacin is a second generation fluroquinolone derivative, exhibiting activity against a wide range of Gram-negative and Gram-positive facultative bacteria as well as periodontal pathogens15 . Diclofenac sodium has analgesic, antipyretic and antiinflammatory activities. It is a non-selective cyclo-oxygenase inhibitor with potency greater than several other NSAIDs. In addition, diclofenac appears to reduce the intracellular concentration of free arachidonate in leucocytes, perhaps by altering the release or uptake of the fatty acid16 . Therefore, an attempt has been made in this study to develop chitosan films containing an antibiotic, ciprofloxacin hydrochloride, and an NSAID, diclofenac sodium, for the treatment of periodontitis. EXPERIMENTAL Materials Ciprofloxacin was a gift from Dr. Reddy’s Laboratory, Hyderabad, India. Diclofenac sodium from Eros Pharmaceuticals, Bangalore, India; chitosan from Central Institute of Fisheries Technology, Kochi, India; and HPLC-grade solvents from Ranbaxy Fine Chemicals Ltd, India, were also used in the study. Preparation of ciprofloxacin-loaded chitosan films Chitosan (2 g) was soaked in 100 ml aqueous acetic acid solution (1% v/v) for 24 hour to get a clear solution, which was later filtered through a muslin cloth to remove undissolved polymer (chitin). Ciprofloxacin (0, 10, 20 and 30 % w/w based on the weight of chitosan) was incorporated in 100 ml of chitosan solution and vortexed (Talboys, Standard Vortex Mixer) for 15 min. The viscous dispersion was kept aside for 30 min for complete expulsion of air bubbles. Films were cast by pouring the drug-polymer solution into the center of glass moulds and allowed to dry at room temperature. The dry films were cut into strips of (7 × 2 mm), wrapped in aluminum foil and stored in a calcium chloride desiccator at room temperature pending evaluation. Preparation of chitosan films containing ciprofloxacin and diclofenac sodium followed the same procedure17 . The compositions of the films are stated in the footnotes of Tables 1 and 2. Preparation of crosslinked chitosan films The films containing 30% drug(s) were crosslinked by exposure to glutaraldehyde vapor in a chromatography chamber. The chamber was previously saturated with the vapour of 2%v/v glutaraldehyde for 24 h; the films were exposed to the vapour for 2 and 4 h, respectively, and then dried. The dry films were cut into strips (7 × 2 mm), wrapped in aluminium foil and stored in a calcium chloride desiccator for further study14 . Evaluation of drug-loaded chitosan films Infra-red (IR) spectroscopy Compatibility studies were conducted, using FT-IR spectroscopy, on the drug(s) alone, the polymer alone and the drug with polymer by potassium bromide cup method. Mixtures of drug alone and drug with polymer were examined under nitrogen to eliminate oxidative and pyrolytic effects at a standard heating rate (2.5 or 10°C/ min). The samples were kept in sealed glass vials for 2 weeks at 60°C and observed for any physical change in appearance. They were again examined by FT-IR spectroscopy18 . Selected film parameters Various film properties such as size, thickness, content uniformity, weight variation and moisture loss were determined. The thickness of three films (1×1 cm) was determined at six different points on the film using a film thickness tester (Mitutoyo 4026F, Japan)15 and the mean calculated16 . Individual weights of twenty films of the same size (7×2 mm) were weighed on an electronic balance and the mean weight was calculated. Percent moisture loss was determined by keeping the film in a desiccator containing anhydrous calcium chloride. After three days, the films were taken out, re-weighed and the percent moisture loss was calculated using the formula: (initial wt – final wt/initial wt) ×100. Tensile strength and elongation Tensile strength was evaluated using an Instron Universal Testing instrument (Model 4206, Instron Ltd., Japan) with a 2 kg load cell. Films of the required dimension and free from air bubbles or physical imperfections were held between two clamps positioned at a distance of 3 cm. During measurement, the top clamp, at a rate of 100 mm/min, was pulled, and the force and elongation were measured when the film broke. Only results from film samples that broke between the clamps were used. Measurements were run in triplicate for each film. Tensile strength and percent elongation were calculated by applying the following equations17 : Tensile strength = Force at break (N)/Initial cross sectional area of the sample (mm2 ) …………..(1) % Elongation = (Increase in length/original length) x 100 ……………………….............................. (2) Estimation of content uniformity The drug-loaded films of known weight (7 × 2 mm) were dissolved in 10 ml of aqueous acetic acid, suitably diluted and the amount of drug(s) present was estimated by UV/VIS spectrophotometer ( Jasco V-530) at 278 nm14 . In vitro drug release studies Since the pH of gingival fluid lies between 6.5 – 6.8, phosphate buffer pH 6.6 was used as simulated gingival fluid. Also, since the film should be immobile in the periodontal pocket, a static dissolution model was adopted for the dissolution studies. Sets of six films of known weight and dimension were placed separately in small sealed test tubes containing 1.0 ml of phosphate buffer (pH 6.6) and kept at 37 ± 0.5 °C for 24 h. The buffer was then drained off and replaced with a fresh 1.0 ml of buffer. The concentration of drug(s) was determined by UV/VIS spectrophotometer (Jasco V-530) at 278 nm and 273 the procedure was continued for 7 and 21 consecutive days for noncrosslinked and crosslinked films, respectively. Following the in vitro release studies, the test films were further analyzed for unreleased drug by dissolving the films in aqueous acetic acid with suitable dilution. The amount of drug released into the dissolution medium plus residual drug content of the films were summed to obtain the actual drug content14 . Scanning electron microscopy (SEM) The morphology and surface topography of the films were examined by SEM (model JSM840 A, SEM–Jeol, Japan). Spherical samples (5 mm2 ) were mounted on the SEM sample stab using a double-sided sticking tape. The samples were coated with gold (200 A0 ) under reduced pressure (0.001 torr) for 2 min using an ion sputtering device (model JFC-1100 E, Jeol, Japan). The gold-coated samples were observed under the SEM and photomicrographs of suitable magnifications were obtained18 . In vitro antibacterial activity In vitro antibacterial activity was performed on all formulations by placing the film, 0.5 × 0.5 cm, on agar plates seeded with the oral bacteria Streptococcus mutans. After 48 h of incubation at 37°C, the films were transferred to freshly seeded agar plates and incubated for an additional 48 h. This procedure was repeated until no inhibition of bacterial growth was detected on the agar plate. The growth inhibition zone on the agar plate was measured19 . Stability studies Three sets (12 strips in each set) of films (size: 7×2 mm) were weighed. The films were wrapped individually in aluminium foil and butter paper (30 GSM, Shree Ganesh Industries, New Delhi) and placed in Petri dishes. These containers were stored at ambient humidity conditions in a refrigerator (Labtop, LLR-290 at 4-8ºC), at room temperature (25 ± 2ºC) and in an oven (Steridium, 140, Australia)at 45 ± 2ºC). The samples were analyzed for physical changes such as color and texture. Drug content was estimated at regular intervals as described earlier and the degradation rate constant, k, was determined by graphical method (log % drug remaining vs time)21 . RESULTS The amount of drug added to the polymer solution altered the film characteristics. The optimum drug loading was less than 30% w/w of the polymer. At higher drug loadings (i.e., > 30%), the films were stiff and brittle. The results of the physicochemical evaluations are presented in Table 1. Moisture loss varied between 4.4 ± 2.2 and 9.9 ± 1.0%. Tensile strength was lowest for uncrosslinked films and highest for crosslinked films and was in the range, 0.541 -1.87 kg/mm2 . In contrast, elongation was greater for uncrosslinked films than for crosslinked films. Drug release data for both uncrosslinked and crosslinked films are illustrated in Figures 1 and 2, respectively. The results showed an initial burst release followed by controlled drug release for up to 7 days for uncrosslinked films (87-95%) and 21 days for crosslinked films (70-78 %). The in vitro antibacterial activity, i.e., 100% inhibition of S mutans was found on day 1 with formulation C-III, while a similar level of antibacterial activity was observed for formulations C-III 2 CL and C-III 4CL on days 3 and 9, respectively. Scanning electron microscopy (SEM) showed differences in the surfaces of the films. The upper surface of blank films and ciprofloxacin films was smooth, while the surface of films containing ciprofloxacin with diclofenac was rough. The degradation rate constant, k, in the stability study was calculated at different temperatures. The values for different formulations ranged from 0.493 to 0.798 under refrigerated conditions while at room and oven temperatures, the values ranged from 0.527 to 1.027 and 0.706 to 1.298, respectively. DISCUSSION The optimum drug loading for good, flexible films was found to be ≤30%. As the concentration of crosslinking agent and/or duration of crosslinking increased, the physical properties of films such as brittleness, tensile strength were altered. The drugs incorporated lowered the tensile strength and elongation of films, indicating that the films had become more brittle. Thus the drugs might have disrupted the linear structures of the polymer chains. Tensile strength was lower for uncrosslinked films than for crosslinked films, due probably to the increased toughness and rigidity of the polymeric film following crosslinking. Conversely, elongation was higher for uncrosslinked than for crosslinked films. Film thickness was satisfactory. Drug content uniformity data suggest good reproducibility of the formulation method used. The uncrosslinked films showed greater moisture loss than the cross-linked films and this may be due to the greater compactness and hence lower porosity of the cross-linked films. In vitro drug release kinetic analysis showed that release mechanism for all the films fitted best to the Highuchi model, as the plots showed high linearity (R2 = 0.913 to 0.9533). The plot of cumulative drug release per unit area versus square root of time in days showed a near linear relationship from 3rd to 7th day and 3rd to 18th day for uncrosslinked and crosslinked films, respectively. This is confirmed by data based on Korsmeyer et al’s equation which showed good linearity (R2 : 0.9566 to 0.9958) and with slope (n) values ranging from 0.176 to 0.304, indicating that zero order diffusion is the prime mechanism of drug release. Mass balance studies indicate that the drug content did not differ from the experimental drug content by more than 3%. Findings in respect of in vitro antibacterial activity show that crosslinked films exhibited antibacterial activity for a longer period (20 days) than uncrosslinked films (7 days). Thus greater crosslinking in crosslinked films resulting in more compactness might have resulted in a more sustained release of drug from the crosslinked films. Scanning electron microscopy showed that the upper surface of plain films was smooth while the surface of films containing ciprofloxacin and diclofenac sodium was rough suggesting that the drug(s) were dispersed rather than dissolved in the polymer solution prior to film formation. The stability results indicate that the films were relatively stable (as reflected by reduced k values) when stored in a refrigerator and at room temperature, compared to those stored in oven temperature conditions. CONCLUSION The advantages of intra-pocket delivery over systemic delivery in periodontitis are that administration is less time-consuming than mechanical debridement and a lower dose of drug would be required to achieve effective therapeutic concentration at the site of action. The drug-loaded chitosan films were flexible, possessed good tensile strength and demonstrated satisfactory physicochemical characteristics. Although the films showed an initial burst release of drug of more than 40%, release was sustained for up to 7 and 21 days for uncrosslinked and crosslinked films, respectively. Thus chitosan films loaded with ciprofloxacin and diclofenac, particularly those crosslinked with gluteraldehyde, may have a role for the therapy of periodontitis. ACKNOWLEDGEMENT We thank Dr. EVS Subrahmanyam, Principal and Dr D Satyanarayana, Research Director, PG and Ph.D studies, NGSM Institute of Pharmaceutical Sciences, Deralakatte, Mangalore, India for providing the required facilities for the research work. We also express our sincere thanks to the staff of the Department of Metallurgy and Material Sciences, National Institute of Technology, Suratkal, Karnataka, India for their cooperation in carrying out some characterization studies. REFERENCES
© Pharmacotherapy Group, Faculty of Pharmacy, University of Benin, Benin City, 300001 Nigeria. The following images related to this document are available:Photo images[pr09006f1.jpg] [pr09006t2.jpg] [pr09006t1.jpg] [pr09006f2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}