 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
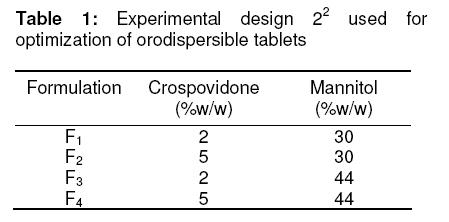
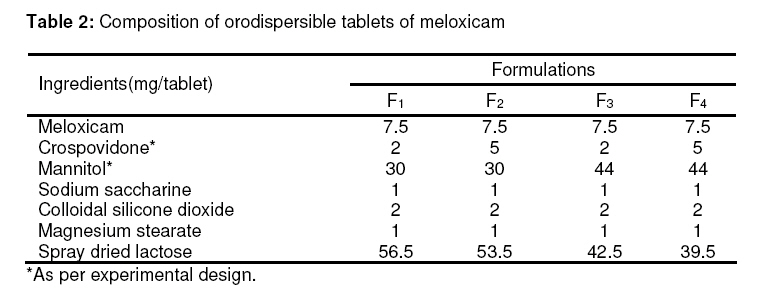
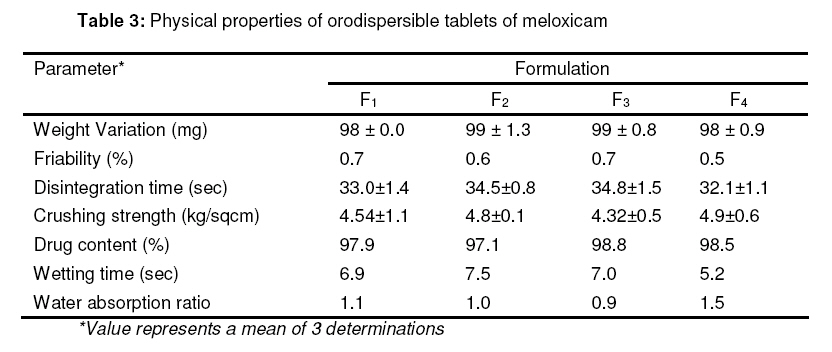
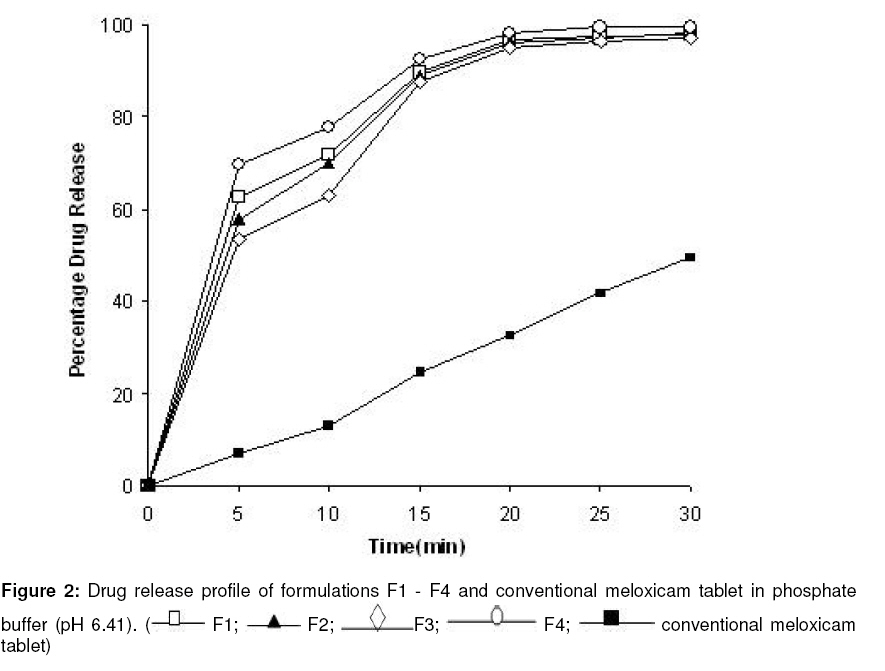
Tropical Journal of Pharmaceutical Research, Vol. 8, No. 2, June 2009, pp. 153-159 Research Article Optimization and Formulation of Orodispersible Tablets of Meloxicam Jashanjit Singh1* and Rajmeet Singh2 1 Department of Pharmaceutics, Swift School of Pharmacy, Village-Gaagar Sarai,Rajpura, Received: 29 September 2008 Revised accepted: 28 December 2008 Code Number: pr09021 Abstract Purpose : The objective of this study was to formulate and optimize an orodispersible formulation of meloxicam using a 22 factorial design for enhanced bioavailability. Keywords: Meloxicam, Orodispersion, Crosspovidone, Mannitol, Factorial design, Tablet properties. Introduction Difficulty in swallowing is a common problem of all age groups, especially geriatric and pediatric patients, due to physiological changes associated with these groups. These problems can be solved by development of a novel type of solid dosage form, namely, orodispersible tablet, which disintegrates and dissolves rapidly in saliva without the need of swallowing with drinking water since the tablet is placed in the mouth where it disperses rapidly before swallowing1,2 . Clinically, nonsteroidal anti-inflammatory drugs (NSAIDs) are the most frequently prescribed by physicians for inflammatory disorders. Meloxicam [4-hydroxy-2methyl-N(5-methyl-2-thiazolyl)-2H-1,2-benzothiazine-3carboxamide1,1-dioxide], a nonsteroidal antiinflammatory drug, was chosen as a model drug for this study. It is one of the most commonly prescribed NSAIDs for the treatment of various inflammatory conditions such as rheumatoid arthritis, osteoarthritis, low back pain3. Although, it has excellent bioavailability (89%)4, its poor aqueous solubility5 makes absorption and dissolution rate-limited, thus delaying onset of action. Consequently, an orodispersible formulation of meloxicam was developed that could enhance the bioavailability and extent of bioabsorption of the drug. Experimental Materials Meloxicam was obtained from Siemen Laboratories, Haryana, India while Crosspovidone was supplied by Signet Chemicals, Mumbai, India. Mannitol, colloidal silicone dioxide and spray dried lactose were obtained from Ranbaxy Fine Chemicals Ltd, New Delhi, India. Sodium saccharine and magnesium stearate were obtained from S.D. Fine Chemicals, Mumbai, India. All other solvents used were of analytical grade. Methods Experimental design The 22 factorial design6 was implemented for the optimization of meloxicam orodispersible tablet. The dependent response measured was disintegration time. Two independent factors -the concentration of crospovidone and concentration of mannitol -were set at two different levels. High and low levels of each factor were coded +1 and –1, respectively (Table 1). Preparation of meloxicam tablets Orodispersible tablets of meloxicam were prepared as per the formula given in Table 2. Accurately weighed quantities of meloxicam, mannitol, crospovidone and other excipients were passed through sieve number 10 (as per Indian Pharmacopoeia)7 and mixed in a glass mortar. The above blend was granulated with a non-aqueous granulating agent, alcoholic solution of PVP (10%w/v), and passed through a sieve of aperture size, 710 µm. The granules were air-dried, mixed with 2%w/w colloidal silicone dioxide, and the granules were passed through sieve number 22, (as per Indian Pharmacopoeia)7 lubricated with magnesium stearate and compressed using CADMACH SMS25 single punch tablet machine at a fixed compression force of 400 kgf . The mean weight and diameter of the tablets were 100mg and 8mm, respectively. Evaluation of orodispersible tablet All the tablets were evaluated for the following parameters: Weight variation Twenty tablets were randomly selected from each batch and individually weighed. The average weight of these selected tablets was calculated. Friability Tablet friability8 was measured using a ROCHE friabilator (USP) at 25 rpm for 4 min. The weight of twenty tablets before and after completion of the test was recorded and friability was calculated by the following formula: Percentage friability= (initial weight-final weight/initial weight)×100 ……………… (1) Disintegration time Three tablets per batch were evaluated for disintegration time by employing a modified dissolution apparatus9 . Instead of the disintegration apparatus described in JP XII, a modified dissolution apparatus (JP XII paddle method) was employed. Water (900 ml), maintained at 37±0.5 °C was stirred with a paddle at 100 rpm. Disintegration time was recorded when all the fragments of the disintegrated tablet passed through the screen of the basket. Crushing strength Tablet crushing strength, which is the force required to break the tablet, was measured with a Pfizer tablet hardness tester. The hardness (crushing strength) of three tablets per batch was determined and mean taken. Percentage drug content Drug content was determined by taking randomly ten tablets per batch. An amount equivalent to 10 mg meloxicam was dissolved in methanol, suitably diluted with 0.1N HCL and filtered. The absorbance of the solution was measured spectrophotometrically against the blank (0.1 N HCl) at 354 nm using a U.V.spectrophotometer (Shimazdu-1700, Japan) Kinetic digital images One tablet of the selected formulation was placed in a glass full of water (see Figure 1) and the dispersion process was recorded, without agitation, and kinetic digital images were taken with a 7.2 mega pixel camera (Sony-DSC-W55, Japan). Wetting time and water absorption ratio The wetting time of the tablet was measured by placing five circular tissue papers (10 cm in diameter) in a Petri dish of 10 cm diameter. Water (10 ml) containing methylene blue (0.1% w/v) was added to the Petri dish. A tablet was carefully placed on the surface of the tissue paper and the time required for the dye to reach the upper surface of the tablet was recorded as wetting time. The measurements were carried out in triplicate. Water absorption ratio was calculated using Eq. 210 : Water absorption ratio = (Wa –Wb )/ Wb ….. (2) where Wb = weight of tablet before absorption of water , and Wa = weight of tablet after absorption of water. In vitro drug release In vitro drug release studies were carried out using USP type II apparatus at 50 rpm. Phosphate buffer (900 ml) at pH 6.41 (corresponding to salivary pH) was used as the dissolution medium. The temperature of the dissolution medium was maintained at 37±0.5 °C. An aliqout (5 ml) of dissolution medium was withdrawn at specific time intervals, filtered and suitably diluted prior to spectrophotometric analysis. Sink conditions were maintained by replenishing the medium with an equal amount (5 ml) of dissolution fluid. Absorption of the solution was measured by UV spectroscopy (Shimadzu-1700, Japan) at 354 nm. Dissolution rate was evaluated for all the formulations (F1 -F4 ) and the conventional tablet. Validation of the experimental design In order to validate the experimental design using polynomial equation, disintegration time was selected. A two-level experimental design provides sufficient data to fit a polynomial equation11 (Eq. 3) which is in the following form: y = B0 + B1 X1 + B2 X2 + B12 (X 1 X2 ) ……(3) where y represents the experimental response, B0 the intercept and B1 to B12 are the coefficients for the factors X1 (crospovidone) and X2 (mannitol). Students ttest was employed to examine the probability of each coefficient being equal to zero. All tests were performed at a 95% confidence level (P>0.05). In the final model equation, only the significant factors were included. The polynomial equation was applied to the response parameter, disintegration time, using a software (New Statistica 10v, USA). Results The results of the measurements of various tablet parameters are tabulated in Table 3. Weight variation and friability The weight range was 98 -99mg for all the tablets. This complies with Indian Pharmacopoeia12 specifications for uniformity of weight, thus indicating consistency in the preparation of the tablets and minimal batch to batch variation. The India Pharmacopoeia prescribes a friability <1% for good mechanical resistance. All the tablet formulations satisfied this requirement as friability was in the range of 0.51 -0.69% Disintegration time All the formulations complied with the dispersion time requirement of ≤60 sec for orodispersible tablets as per European Pharmacopoeia13. Formulation F4 had the least dispersion time of 32.1 sec (Table 3). Crushing strength The hardness of the formulated tablets was in the range of 4.54 -4.93 kg/cm2 and did not show any significant difference among the various formulations. Wetting time and water absorption ratio The wetting time of all the tablet formulations was within the range of 5.15-7.50 sec and this indicates rapid absorption of water by the tablets due to the wicking action of crosspovidone. The water absorption of formulation F4 was highest with a value of 1.5, indicating a high amount of water uptake due to the high concentration of the independent factors. In vitro drug release All the formulations showed optimized drug release, with 98%-99% of the drug released within 30 min. Validation of the experimental design The theoretical response obtained from the polynomial equation at Y50% was found to be 37.22sec and the experimental response of the extra design point formulation was found to be 34.6sec±0.43 which was quite close to the theoretical response. Discussion Since mannitol has good aqueous solubility, negative heats of solution and good wetting properties, these attributes improve the binding of the tablets and water uptake, thereby decreasing disintegration time. All the formulations disintegrated in less than 60 sec. Formulation F4 demonstrated a minimum dispersion time of 32.1 sec; in this formulation, mannitol (an orodispersion aid) and crosspovidone were at their highest levels. The in vitro disintegration behavior, without agitation, of formulation F4 indicate that complete dispersion was obtained within 40 sec (Figure 1). The difference between this value and the disintegration time obtained by the previous method may be attributed to the agitation involved in the previous method. There was no significant difference (P<0.05) among the four tablet formulations (F1 – F4 ) in terms of their physicochemical properties – disintegration time, crushing strength, friability and wetting time – except for water absorption ratio where F4 clearly showed a higher value than the other tablet formulations. The crushing strength values of 4.54 -4.93 kg/cmare satisfactory and may have been facilitated by the incorporation of 2%w/w colloidal silicon dioxide which improves the hardness of the tablets, since colloidal silicon dioxide is known to enhance the bonding property of excipients14. This also would have had a positive effect on the friability of the tablets as friability was within the limit of 1%. The water uptake ratio of F4 tablets was 1.521 times its actual weight, thus facilitating the process of orodispersion. All the formulations showed good drug release which was in the range, 98% -99%, after 30 min. Thus, incorporation of varying concentrations of crospovidone and mannitol did not significantly alter drug release (Figure 2). However, conventional meloxicam tablets exhibited approximately 50% of the release rate of the optimized tablets (F1 -F4). The formulation followed first order kinetics as a linear plot was obtained when log drug release was plotted against time. The correlation coefficient of the plot was 0.9792. The disintegration time data can be described by a model equation (Eq 4): Ydisintegration time = 28.91+1.21(X1 ) + 2.48(X2 ) +4.62(X1 X2 ) ……………………………….. (4) The positive coefficient for equation X1 indicates that a higher level of crosspovidone in the tablet formulation facilitated disintegration. The theoretical response at Y50% was 37.22sec while the experimental response of the extra design point formulation was 34.6sec±0.43 which was quite close to the theoretical response, thus validating the experimental design. Conclusion All the optimized tablet (orodispersible) formulations (F1 -F4 ) showed disintegration times that were less than 60 sec, as well as good physicochemical properties. Drug release rates of the orodispersable tablets were much higher than that of the conventional tablets. The experimental design was also validated, as the theoretical response obtained correlated well with the practical response. The results indicate that the presence of crospovidone and mannitol not only enhanced the rate of orodispersion but also improved drug release rate. Thus, satisfactory orodispersible tablets of meloxicam for large-scale production is feasible. References
© Pharmacotherapy Group, Faculty of Pharmacy, University of Benin, Benin City, 300001 Nigeria. The following images related to this document are available:Photo images[pr09021f2.jpg] [pr09021t3.jpg] [pr09021t2.jpg] [pr09021f1.jpg] [pr09021t1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}