 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
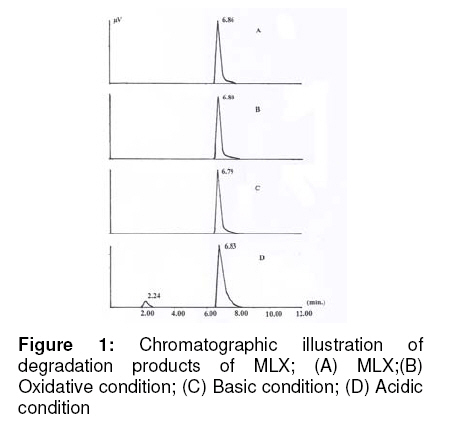
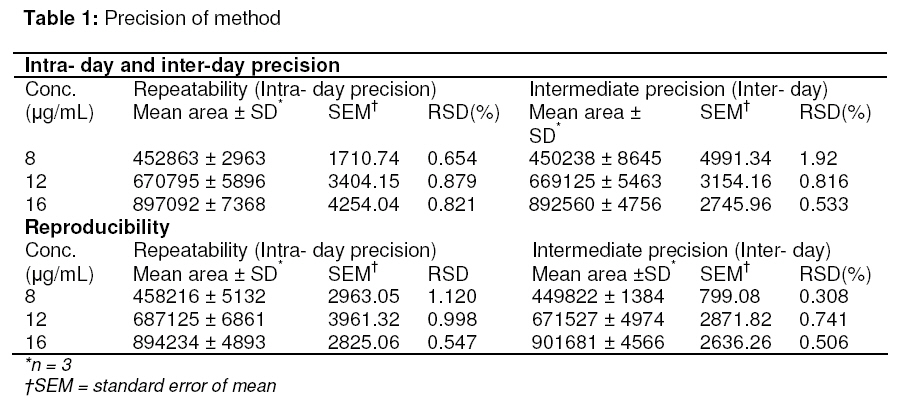
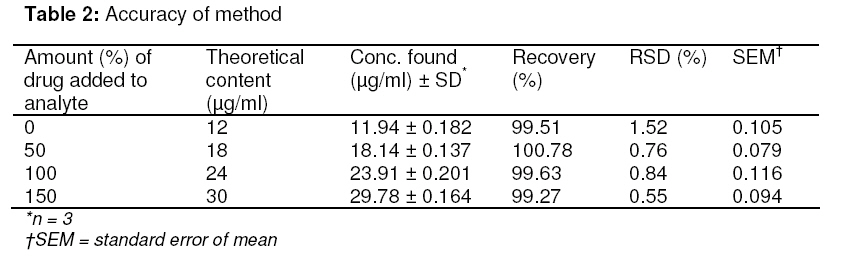
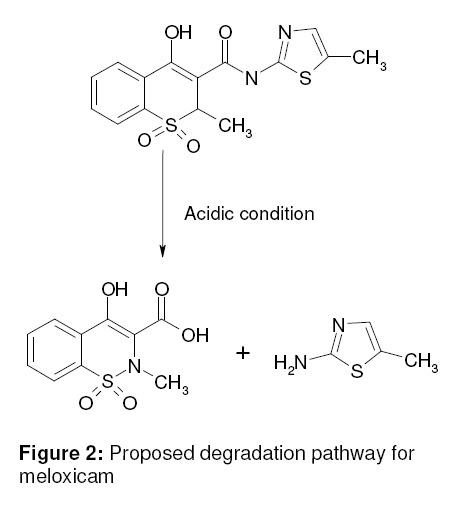
Tropical Journal of Pharmaceutical Research, Vol. 8, No. 3, June 2009, pp. 257-264 Research Article A Stability Indicating HPLC Method for the Determination of Meloxicam in Bulk and Commercial Formulations Farzana S Bandarkar1* and Pradeep R Vavia2 1Department of Pharmaceutics, Faculty of Pharmacy, Kuwait University, Kuwait, Received: 23 Nov 2008 Revised accepted: 3 Mar 2009 Code Number: pr08034 Abstract Purpose: The present study was undertaken to develop a validated, rapid, simple and economic stability indicating reverse phase HPLC method for estimating meloxicam (MLX) in bulk and commercial preparations. Key words: Meloxicam, Reverse phase stability indicating HPLC, Stress degradation, Bulk and commercial formulations. INTRODUCTION Meloxicam (MLX), is an oxicam derivative and a member of the enolic acid group of non-steroidal anti-inflammatory drugs (NSAIDs)1,2. It is chemically designated as 4hydroxy-2-methyl–N-(5-methyl–2–thiazolyl) 2H -1,2-benzothiazine–3-carboxamide-1,1dioxide. MLX exhibits anti-inflammatory, analgesic and anti-pyretic activities, especially in various chronic conditions, like osteoarthritis, rheumatoid arthritis, pauciarticular and polyarticular course juvenile rheumatoid arthritis3,4 . The mechanism of action of MLX is believed to be due to inhibition of prostaglandin synthesis, primarily via inhibition of cyclooxygenase-2 (COX-2). In contrast with other NSAIDs, it has neither acute nor chronic gastrointestinal toxicity5. Various analytical techniques viz, UV spectrophotometry6-8, fluorimetry6, capillary electrophoresis9, pulse polarography10, electrochemical oxidation11, electrochemical reduction12 and voltametry13 are reported for the analysis of MLX in pharmaceuticals. HPLC is the most commonly used method for analysis of MLX. An extensive literature survey reveals few HPLC methods for estimation of MLX in pharmaceutical dosage forms as well as biological fluids; however, not all of these are stability indicating and some of them make use of buffer in the mobile phase. Most of the reported methods either do not include stress degradation studies or are not completely validated, and they are cumbersome, time-consuming and expensive14-17. Method validation is an essential step in drug analysis. The process confirms that the analytical procedure employed for the analysis is suitable for its intended use and shows reliability of the results produced by any method. The primary objective of the present work was thus to develop and validate a stability indicating HPLC method for MLX, which could also be employed for the routine analysis of the drug in pharmaceutical dosage forms. In the method proposed, the mobile phase was used directly for the dilution of the formulation after filtration, and then further used for analysis. Direct use of the mobile phase as diluent for formulations in quantitative analysis minimizes errors that occur during tedious extraction procedures. The method was validated in accordance with ICH guidelines18. EXPERIMENTAL Materials MLX was received as a gift from Unichem Lab. Ltd., Mumbai, India. Acetonitrile (HPLC – grade) was purchased from Merck, India. Millipore purification system was used for high purity water. All other chemicals and reagents employed were of analytical grade and were purchased from S.D. Fine Chemicals, India. Chromatography method The chromatograph system comprised of a Jasco PU-980 pump equipped with a Jasco UV-975 detector and a Rheodyne injector with a 20-microlitre loop. Data integration was done using a Borwin software package V1.21. Samples were injected into a Hi-Q-Sil C-18 column (4.6 x 250mm, 5 µ particle size). Mobile phase flow rate was 1ml/min. The drug was analyzed at a wavelength of 355nm. Method development Initial trial experiments were conducted, with a view to select a suitable solvent system for the accurate estimation of the drug and to achieve good resolution between the drug and the degradation products. The suitability of the mobile phase was decided on the basis of the sensitivity of the assay, suitability for stability studies, time required for the analysis, ease of preparation, and use of readily available cost-effective solvents. These included methanol–water (50:50 % v/v), acetonitrile–water, (50:50 % v/v), acetonitrile-water (60:40 %v/v), acetonitrilewater-glacial acetic acid (54:44:2 % v/v) and acetonitrile-water-glacial acetic acid (55:40:5 % v/v). A mobile phase system comprising of acetonitrile-water-glacial acetic acid (55:40:5 % v/v) was found to be optimum. The same solvent mixture was used for the extraction of the drug from the formulation containing excipients. The solvents were mixed, filtered through a membrane filter of 0.45 micron pore and degassed before use. Method validation Linearity A series of standard curves were prepared over a concentration range of 4 -20 µg/ml from a stock solution of MLX (1mg/ml) in acetonitrile. Dilutions were prepared in the mobile phase: acetonitrile-water-glacial acetic acid (55:40:5 %v/v). The procedure for analysis follows that described earlier under the subsection, ‘Chromatography method’. The data from peak area versus drug concentration plots were treated by linear least square regression analysis. The standard curves were evaluated for intra-day and inter-day reproducibility. Each experiment was repeated in triplicate. Precision Precision is the measure of how close the data values are to each other for a number of measurements under the same analytical conditions. The three components of precision, i.e., repeatability, intermediate precision and reproducibility, in accordance with ICH recommendations, were determined as follows: Repeatability Injection repeatability: Five injections of 12 µg/mL solution of MLX were analyzed and %RSD calculated for injection repeatability. Intra-day variation: Measurement of intra-day variation of MLX solutions at three different concentrations (8, 12 and 16 µg/mL) was carried out by injecting the samples on the same day at different time intervals. Analysis repeatability: It was obtained by determining the relative standard deviation (RSD) of replicate samples (n=3) of the accuracy study. Intermediate precision (Inter-day variation) Measurement of inter-day variation of MLX solutions at three different concentrations (8, 12 and 16 µg/mL) in triplicate on three consecutive days determined the intermediate precision. Reproducibility The reproducibility of the method was checked by determining precision on the same instrument, but by a different analyst. For both intra-day and inter-day variation, solutions of MLX at three different concentrations (8, 12, and 16 µg/mL) were analyzed in triplicate. Accuracy Accuracy is the measure of how close the experimental value is to the true value. Recovery studies by the standard addition method were performed with a view to justify the accuracy of the proposed method. Previously analyzed samples of MLX (12 µg/ml) were spiked with 50, 100, and 150% extra MLX standard and the mixtures were analyzed by the proposed method. The experiment was performed in triplicate. Recovery (%), RSD (%) and standard error of mean (SEM) were calculated for each concentration. LOD and LOQ In order to estimate the limit of detection (LOD) and limit of quantitation (LOQ) values, the blank sample was injected six times and the peak area of this blank was calculated as noise level. The LOD was calculated as three times the noise level while ten times the noise value gave the LOQ. Robustness The robustness of the method was determined to assess the effect of small but deliberate variation of the chromatographic conditions on the determination of MLX. Robustness was determined by using reagents from two different lots and two different manufacturers. Sample solution stability The stability of the drug in solution during analysis was determined by repeated analysis of samples during the course of experimentation on the same day and also after storage of the drug solution for 72 h under laboratory bench conditions (25 ± 1 °C) and under refrigeration (8 ± 0.5 °C). An accurately weighed quantity of the pure drug was dissolved in acetonitrile and suitably diluted with mobile phase to get a final concentration of 12 µg/ml. The solution was subjected to HPLC analysis immediately and after a period of 24, 48 and 72 h. Specificity/Selectivity The specificity of the method was determined by exposing the sample solution (12 µg/mL) to acidic (0.1 M HCl), basic (0.1 M NaOH), and oxidising (3% H2 O2 ) stress conditions. The samples were refluxed for 10 h at 100ºC, filtered and analyzed. System suitability tests The chromatographic systems used for analyses must pass the system suitability limits before sample analysis can commence. The capacity factor (K), injection repeatability (as described earlier in the subsection, ‘Precision’), tailing factor (T), theoretical plate number (N) and resolution (Rs) for the principal peak and its degradation product were the parameters tested on a 12 µg/mL sample of MLX to assist the accuracy and precision of the developed HPLC system. Analysis of MLX in marketed tablets Ten tablets (strength: 15 mg/tablet) were crushed and triturated well in a mortar. A powder sample, equivalent to 15mg of MLX, was accurately weighed and transferred to a 25ml volumetric flask. The drug was extracted into acetonitrile and mixed thoroughly for 30 min using a sonicator. The solution was filtered through 0.45 micron pore filter after making up the volume, adequately diluted with mobile phase and analyzed by the proposed HPLC method. The possibility of interference of excipients with the analysis was studied. Data analysis The r value for the calibration plot, SD, RSD, and SEM were determined using Microsoft Excel 2007 application. RESULTS Method development Acetonitrile-water-glacial acetic acid (55:40:5 %v/v) was selected as the optimum mobile phase. Under these conditions the retention time and tailing factor were 6.8 ± 0.01 min and 1.13 respectively. A typical chromatogram is represented in Fig. 1A. Method validation Linearity Peak area versus drug concentration was plotted to construct a standard curve for MLX. The polynomial regression for the calibration plots showed good linear relationship with coefficient of correlation, r = 0.9995 ± 0.0092; slope = 57257.38 ± 165.74 and intercept = 3443.07 ± 97.56 (n = 6) over the concentration range studied. The range of reliable quantification was set at 4 – 20 µg/ml as no significant difference was observed in the slopes of the standard curves in this range. The linear regression data for the calibration plot is indicative of a good linear relationship between peak area and concentration over a wide range. The correlation coefficient was indicative of high significance. The low values of the standard deviation, the standard error of slope, and the intercept of the ordinate showed the calibration plot did not deviate from linearity. Precision Precision was measured in accordance with ICH recommendations. Five consecutive injections of 12 µg/mL solution of MLX by the proposed method showed excellent injection repeatability with RSD of only 0.47%. Repeatability of sample injection was determined as intra-day variation while intermediate precision was determined by measuring inter-day variation for triplicate determination of MLX at three different concentrations. The results of the determination of repeatability, intermediate precision and reproducibility are listed in Table 1. Reproducibility was checked by measuring the precision of the proposed method with analysis being performed by another person. The low RSD values indicate the repeatability and reproducibility of the method. Recovery The recovery of the method, determined by spiking a previously analyzed test solution with additional drug standard solution, was found to be in the range of 99.27 – 100.78%. The values of recovery (%), RSD (%) and SEM listed in Table 2 indicate the method is accurate. Detection and Quantification limits The limit of detection was found to be 360 ng/ml where the drug could be detected without any noise. The limit of quantification was 510 ng/ml. This indicated the method can be used for detection and quantification of MLX over a very wide range of concentrations. Robustness There was no significant change in the retention time of MLX when reagents (acetonitrile and glacial acetic acid) from different lots and different manufacturers were used. The concentration of the solution analyzed was 12 µg/mL and the % RSD ranged from 0.078 to 1.286 %. The low values of the RSD indicated the robustness of the method. Stability There was no significant change in analyte composition (sample concentration = 12 µg/mL) over a period of 72 h. The mean RSD between peak areas, for the samples stored under refrigeration (8 ± 1°C) and at laboratory temperature (25 ± 1°C) was found to be 0.990% and 0.771% respectively, suggesting that the drug solution can be stored without any degradation over the time interval studied. Specificity The specificity of the method was determined by exposing 12 µg/mL sample solutions of MLX to stress conditions, i.e., 0.1 M HCl, 0.1 M NaOH, and 3% H2 O2 . There was no degradation of MLX in the presence of 0.1 M NaOH or 3% H2O2 and no significant change in peak area and retention time of MLX was observed (Fig 1). However, in the presence of 0.1N HCl, it was found that there was a substantial change in the peak area of MLX, but not in the retention time. Chromatograms obtained from MLX after treatment with 0.1 M HCl, 0.1 M NaOH, and 3% H2 O2 are shown in Fig 2. A degradation product (Fig 1D) eluted with a retention time of 2.24 ± 0.03 min. The results from stress testing, including separation of the degradation product and quantification of MLX after exposure to stress conditions, show that the method is stabilityindicating. System suitability tests The results of the system suitability tests assure the adequacy of the proposed HPLC method for routine analysis of MLX. The capacity factor (k) was found to be 1.86, indicating that the MLX peak is well resolved with respect to the void volume. The RSD of five consecutive injections performed under the precision test was found to be 0.47% and thus shows good injection repeatability. The tailing factor (T) for MLX peak was found to be 1.13, reflecting good peak symmetry. The resolution (Rs) for the principle peak and its acid degradation product was found to be 6.22, indicating good separation of the drug from its degradation product. The theoretical plate number (N) was found to be 2466, thus demonstrating good column efficiency. Analysis of MLX from marketed tablets A single peak was observed at the retention time of MLX when a suitably diluted solution of the tablet formulation was chromatographed. No interaction was observed between MLX and excipients present in the tablets. The MLX content was found to be 99.46% and the RSD was 0.94%. The low RSD indicated the suitability of this method for routine analysis of MLX in pharmaceutical dosage forms. DISCUSSION The final decision on mobile phase composition and flow rate was made on the basis of peak shape, peak area, tailing factor, baseline drift and time required for analysis. The solvent system selected [acetonitrilewater-glacial acetic acid (55:40:5 %v/v)] gave good resolution of degraded product and drug peak. No internal standard was used because no extraction or separation step was involved. Methanol-water (50:50 %v/v) did not furnish a sharp, well-defined peak and but effected a high tailing factor (1.82). Other mobile phases tried resulted either in much lower sensitivity, delayed retention time or poor peak shapes, and so were not considered. The proposed HPLC method of analysis was also found to be precise and accurate, as depicted by the statistical data of analysis. High values of correlation coefficients and small values of intercepts validated the linearity of the calibration plots and obedience to Beer’s laws. The RSD values and the slopes and intercepts of the calibration graphs indicate the high reproducibility of the proposed method. The method was also found to be robust as there was no significant change in the peak area, peak shape and retention time of MLX. Furthermore, the low values of LOD and LOQ indicate that the method can be employed over a wide concentration range for linearity. This method is also highly sensitive and could effectively separate the drug from its degraded product. MLX is a thiazolyl substituted benzothiazine carboxamide. Solution of MLX is stable at room temperature and when refluxed at 100ºC for 10 h with a strong base (NaOH) or hydrogen peroxide solution. However, when refluxed at 100ºC with a strong acid (HCl) for 10h, hydrolysis of the amide group takes place, resulting in the formation of the corresponding carboxylic acid and amine (Rt of 2.24 min.). Fig 2 represents the proposed degradation pathway. As the reported method could effectively separate the drug from its degraded product, it can be employed as a stability indicating one. The system suitability tests performed verified the resolution, column efficiency and repeatability of the chromatographic system and ensured that the equipment, electronics, and analytical operations for the samples analyzed could be constituted as an integral system that can be evaluated as a whole. CONCLUSION The HPLC method developed is accurate, precise, reproducible, specific, and stabilityindicating. The method is linear over a wide range, economical and utilizes a mobile phase which can be easily prepared. All these factors make this method suitable for quantification of MLX in bulk drugs and in pharmaceutical dosage forms. It can therefore be concluded that use of the method can save much time and money and it can be used even in small laboratories with very high accuracy and precision. The method can also be used for the routine analysis of MLX in bulk preparations of the drug and in pharmaceutical dosage forms without interference. ACKNOWLEDGEMENT The authors are thankful to Unichem Laboratory Ltd., Mumbai, India, for the gift of MLX. REFERENCES
© Pharmacotherapy Group, Faculty of Pharmacy, University of Benin, Benin City, 300001 Nigeria. The following images related to this document are available:Photo images[pr09034t2.jpg] [pr09034f1.jpg] [pr09034f2.jpg] [pr09034t1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}