 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Tropical Journal of Pharmaceutical Research, Vol. 8, No. 5, October, 2009, pp. 425-432 Research Article Effect of Hydroxypropyl Methylcellulose and Ethyl Cellulose Content on Release Profile and Kinetics of Diltiazem HCl from Matrices R Enayatifard, M Saeedi, J Akbari and Y Haeri Tabatabaee Department of Pharmaceutics, Faculty of Pharmacy, Mazandaran University of Medical Sciences, Sari, Iran *Corresponding author: E-mail: enayatifard_r@yahoo.com; Tel: +98-151-3543082; Fax: +98-151-3543084 Received: 27 February 2009 Code Number: pr09055 AbstractPurpose: To develop a oral controlled matrix drug delivery
system for a highly water soluble drug, diltiazem HCl, and investigate its drug
release mechanism. Key words: Matrix tablets; Diltiazem HCl; Hydroxypropyl methylcellulose; Ethylcellulose; Drug release INTRODUCTIONOral ingestion has long been the most convenient and commonly employed route of drug delivery due to ease of administration, high patient compliance, least sterility constraints and flexibility in the design of dosage form. Hydrophilic polymers are becoming increasingly popular in the formulation of oral controlled release tablets. As the dissolution medium or biological fluid penetrates the dosage form, the polymer material swells and drug molecules begin to move out of the system by diffusion at a rate determined by the nature and composition of the polymer as well as formulation technology [1]. Cellulose ethers, especially hydroxyl-propyl methylcellulose (HPMC), are frequently used as the basis for sustained release hydrophilic matrix tablets. Despite studies in the 1960s describing their uses [2,3], their characterization and performance have been more extensively quantified only recently. Their properties as gelling agents are very important in the formulation because they are responsible for the formation, by hydration, of a diffusion and erosion-resistant gel layer which is able to control drug release [4]. On the other hand, hydrophobic materials have also been employed as matrix carriers for sustained release solid dosage forms [5,6]. Ethyl cellulose (EC) is a non-toxic, stable, compressible, inert, hydrophobic polymer that has been widely used to prepare pharmaceutical dosage forms. The properties of ethyl cellulose sustained release products, including film coated tablets [7], microspheres [8,9], microcapsules [10] and matrix tablets for both soluble and poorly soluble drugs [11] have been reported. The combination of ethyl cellulose and a hydrophilic component such as HPMC offers a flexible system to tailor the drug release by changing the viscosity, substitution type and concentration of HPMC [12]. In general, designing controlled release drug delivery systems for providing 12 or 24 h zero order release kinetics, especially for highly water-soluble agents, is often difficult and unsuccessful. This shortfall in delivery system design may be attributed to three factors: (a) the high water solubility of the drug results in a burst effect; (b) the lack of proper control over time-dependent processes of polymer relaxation and disentanglement in relation to drug dissolution and diffusion; and (c) compensation for increase in the diffusional path length with time is not easily achievable [13]. For drugs with high water solubility, hydrophobic polymers are suitable, along with a hydrophilic matrix, for developing sustained release dosage forms. Hydrophobic polymers provide several advantages, ranging from good stability at varying pH values and moisture levels to well-established safe applications. Diltiazem HCl, an orally active calcium channel blocking agent, is used in the treatment of angina pectoris, hypertension and arrhythmia. It is highly water-soluble drug, and is rapidly and almost completely (60-70%) absorbed from GIT following oral administration, but undergoes extensive hepatic metabolism. The biological half-life of the drug is 3.5 ± 1.2 h. It is typically administered three or four times daily, in the form of conventional tablets [14]. To minimise frequency of administration and the peak-to-trough oscillation of the blood concentration of such a typically water-soluble, diltiazem HCl was selected as a suitable candidate for this study [15]. Consequently, this work sought to develop a sustained-release diltiazem hydrochloride system, employing hydrophilic (HPMC) and hydrophobic (EC) polymers either alone or as a blend. Possible mechanisms of the steady-state release kinetics are also discussed. EXPERIMENTAL Materials Diltiazem hydrochloride and ethylcellulose (20cps) were received as gifts from Aria Pharmaceutical Company, Tehran, Iran. HPMC K4M was also a gift from Colorcon, UK While magnesium stearate was purchased from Merck Company,,Germany. Formulation and preparation of matrices In all formulations, 90 mg of diltiazem hydrochloride per tablet was employed and the formulations were prepared according to the composition shown in Table 1. The drug and polymers in their specified ratios were simultaneously blended in a laboratory-scale Z-shape blender for 15 min. The resulting powders were mixed with magnesium stearate for 5 min, following which tablets were prepared from the blends by direct compression on a single punch tablet compression machine (Korsch, Germany) using flat-faced punches with a diameter of 0.9 cm. The compression force was adjusted to achieve maximum hardness. Table1: Composition of 90 mg diltiazem tablet formulations
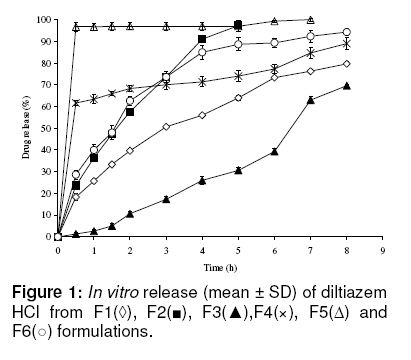
*Formulations code Determination of hardness of matrix tablets The hardness of the tablets were measured using a hardness tester (Erweka TBH-30, Germany) 24h after compaction. Ten (10) randomly selected tablets per batch were measured and the mean taken. Determination of tablet friability The friability of the tablets was determined using 10 tablets from each formulation, with a friabilator (Erweka TAR-20 Germany) at a speed of 25 rpm for 4 min. The tablets were weighed before and after the friability test, and friability was determined as percent weight change. Drug content Ten randomly selected tablets from each formulation were weighed and powdered. The quantity of powder equivalent to 90 mg of diltiazem hydrochloride was transferred into a 100 ml volumetric flask and extracted with distilled water. A quantity (1 ml) of the filtered solution was diluted to 50 ml with distilled water and absorbance was measured at 236 nm (Spectronic Genesys 2, USA). Each measurement was carried out in triplicate and the mean taken. Drug concentration was calculated from a calibration curve based on a standard concentration range of 0 to 20 µg/ml. Dissolution studies In vitro dissolution studies were carried out using a tablet dissolution tester (Erweka, DT 800 Germany). The USP basket method I was used. Rotation speed was 100 rpm and the dissolution medium was distilled water (900 ml) maintained at 37 ± 0.5 oC. Drug release was monitored by UV spectrophotometry (Spectronic Genesys 2, USA) at a wavelength of 236 nm. The test was carried out in triplicate and the results expressed as mean ± standard deviation (SD). Analysis of drug release kinetics The release data were analyzed with the following release models: zero order (Eq. 1), first order (Eq. 2), Higuchi (Eq. 3), and Peppas and Korsemayer (Eq. 4). Qt = k0 t ……………………. (1) Ln Qt = ln Q0 - k1t …….…………….. (2) Qt = kH t1/2 ……………………………………(3) Mt / M∞= Ktn …………………….…(4) Where Qt is the amount of drug released in time t, Q0 is the initial amount of drug in the tablet, Mt corresponds to the amount of drug released in time t, M∞ is the total amount of drug that must be released at infinite time, K is a constant and n is the release exponent indicating the type of drug release mechanism. If n approaches 1, the release mechanism is zero order; on the other hand, if 0.5 < n < 1, non-Fickian transport is the case. The drug release data obtained were subjected to the above drug release models in order to establish their release mechanism and kinetics. Criteria for selecting the most appropriate model were based on best fit and smallest sum of squared residuals. Statistical analysis A one–way ANOVA with a Tukey’s post hoc test was used to analyze the dissolution data obtained for each batch of formulation in order to compare the rate of drug release from the matrix tablets. The confidence limit was set at 95%. RESULTS Table 2 indicates the results of the physicochemical tests (hardness, friability and assay) performed on the tablet formulations. The data show that hardness ranged from 18.14 to 141.74N while friability ranged from 0.16 to 1.61 %. The results of the dissolution studies, shown in Figs 1 and 2, indicate that while formulations F1, F2 and F3 released 36.7, 73.7 and 10.7 % of diltiazem, respectively, after 2 h and 79.7, 100.0 and 69.7 % of drug, respectively, after 8 h, formulations F4, F5 and F6 released 68.3, 96.3 and 62.7 % of diltiazem, respectively, at the end of 2 h, and Table 2: Physical properties of diltiazem HCl matrix tablets
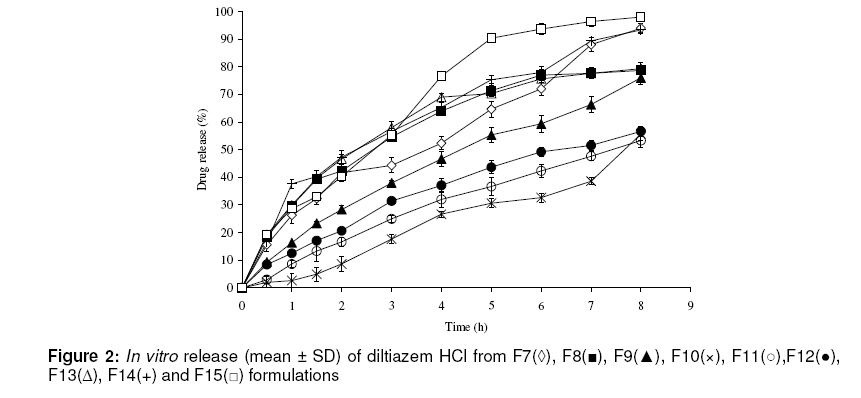
*Formulations code; aMean ± SD, n= 10; bMean ± SD, n= 3 89.0, 100.0 and 100.0 % of drug, respectively, after 8 h (see Fig 1). The dissolution profiles of diltiazem tablets containing blends of HPMC and EC are shown in Fig 2. The tablets with a constant amount of HPMC but varying ratios of EC (F1, F7 and F9) released 79.7, 94.0 and 76.0 % of diltiazem, respectively, after 8 h.
Table 3: Computed kinetic release parameters for diltiazem formulations
F*: code of formulations; K0, the zero order release rate constant, K1, the first order release rate constant; K H, the Higuchi’s release rate constant; KK p, Korsmeyer-Peppas release constant, n, diffusional exponent The results of the analysis of drug release kinetics based on various models are shown in Table 3. It is evident that formulations F3, F7 and F15 followed a zero-order release pattern while formulations F10, F11 and F14 formulations followed first order release, with regression values between 0.9779 and 0.9984. DISCUSSION Physical properties The present study was carried out to develop oral controlled release tablet dosage form for highly water-soluble drugs such as diltiazem hydrochloride using a blend of HPMC and EC as the matrix. As shown in Table 2, the drug content of all formulations was between 97.4 and 99.4 %, indicating the presence of an acceptable amount of drug in the formulations. Furthermore, all the formulations showed acceptable hardness and friability. In vitro drug release The in vitro drug release data obtained over a period of 8 h indicate that for F1, F2 and F3 formulations, the release rate decreased as the concentration of HPMC increased. The results of this study are consistent with findings in a previous report by Pham and Kee [16], which showed that the presence of a highly water-soluble compound, fluorescine, in a HPMC matrix generates an additional osmotic gradient, thereby resulting in a faster rate of polymer swelling and a large increase in gel thickness. In the presence of a solvent, the mobility of the polymer chains is enhanced, resulting in a gradual transformation of a glassy matrix to a rubbery swollen gel. At higher polymer loading, the viscosity of the gel matrix is increased which results in a decrease in the effective diffusion coefficient of the drug [17]. Wan et al have also reported that other factors that may contribute to differences in drug dissolution profile as a function of changes in total polymer concentration include differences in water penetration rate, water absorption capacity and polymer swelling [18]. Incorporation of varying concentrations of ethyl cellulose (F4, F5 and F6) controlled drug release. This may be attributed to decreased penetration of the solvent molecules in the presence of the hydrophobic polymer, leading to reduced diffusion of the drug from the matrix. According to penetration theory, when a matrix is composed of a water-soluble drug and a water-insoluble polymer, drug release occurs by dissolution of the active ingredient through capillaries composed of interconnecting drug particle clusters and the pore network [19,20]. As drug release continues, the interconnecting clusters increase the pore network through which interior drug clusters can diffuse with more ethyl cellulose particles present, and the theory predicts that fewer clusters of soluble drug substance are formed. Furthermore, the presence of finite drug clusters is more statistically plausible. The resulting pore network becomes less extensive and more tortuous resulting in slower drug release. In tablets which were prepared with the same amount of HPMC but varying concentration of EC (F1, F7 and F9), the release rate of diltiazem decreased significantly (P = 0.023) with increasing EC content. Although, incorporation of EC controlled drug release to some extent, the inclusion of this polymer in formulation F7 increased the release of the drug. The reason might be that its large hydrophobic molecules imposed a discontinuity in the gel-structure leading to formation of a weaker barrier than the HPMC gel alone [21]. During the formulation development process, formulation F2 was modified by the addition of EC in varying concentrations to yield F8 and F14. F2, F8 and F14 released 100.0, 78.7 and 93.3% of diltiazem, respectively, within 8 hours. This decrease was significant for F8 and F14 (P ≤ 0.001). Drug release (over 8 h) from EC matrix tablets with varying concentrations of HPMC (F5, F9 and F11) was 100.0, 76.0 and 53.3%, respectively. Thus there were significant differences in drug release between these formulations (P ≤ 0.001). It seems that increasing the concentration of HPMC decreased the release rate of diltiazem, due to the fact that HPMC forms a strong viscous gel on contact with aqueous media with the gel controlling delivery of the highly water-soluble drug. Usually, water-soluble drugs are released primarily by diffusion of dissolved drug molecules across the gel layer [22]. The extent of polymer swelling and the hydration of the microstructure formed within the gel layer vary with the degree of polymer interaction with hydrating media. Drug release kinetics When the release data were subjected to first-order, Higuchi, Korsmeyer et al and zero order models, F10, F11 and F14 formulations showed linearity with regression values between 0.9779 and 0.9984 for first order. Release of drug from a matrix tablet containing hydrophilic polymers generally involves factors of diffusion. The relaxation and swelling characteristics of HPMC matrices may influence drug release kinetics. These matrices have been shown to expand predominantly in an axial direction. Drug release from swelling matrices is dependent on the diffusion and relaxation behavior of the dosage form. The diffusional release is by molecular diffusion down a chemical potential gradient whereas relaxational release is by drug transport mechanisms associated with stresses and state transitions involved in the swelling of the hydrophilic polymer. The swelling of the polymer would alter the drug concentration gradient in the gel layer [23]. As the gradient varies, the drug is released, and the distance for diffusion increases. This could explain why the drug diffuses at a comparatively slower rate as the distance for diffusion increases, which is governed by square-root or Higuchi kinetics. To confirm the release mechanism, the data were fitted into Korsmeyer et al model. F1, F2, F6 and F13 showed good linearity (r2 = 0.9648 to 0.9995), with the slope or exponential value (n) ranging from 0.5802 to 0.6748, indicating a coupling of diffusion and erosion mechanisms – so-called anomalous diffusion. When plotted according to Korsmeyer et al model, F9 also showed high linearity (r2 = 0.9967), with a comparatively high slope (n) value of 0.7442 This n value, however, appears to indicate that diffusion is the dominant mechanism of drug release for this formulation. The in vitro release profile of drug from formulation F9 could be best expressed by Higuchi’s equation, as the plot showed high linearity (r2 = 0.9973). The drug release kinetics of formulations F3, F7 and F15 fitted best to the zero order model. In these formulations the amount of HPMC played a dominant role. CONCLUSION The findings of the present study demonstrate that HPMC and EC could slow down the release profile of diltiazem HCl from their matrices. Incorporation of EC in HPMC matrix tablets was found to control drug release. Release kinetics analysis showed that drug release from three formulations was adequately described by zero-order equation. This approach to formulation development may be suitable for controlled delivery of highly soluble drugs such as diltiazem HCl. ACKNOWLEDGMENT This work was supported by a grant from the Research Council of the Mazandaran University of Medical Sciences, Iran. We would like to thank Dr. Khorrami (Targolshimi Co.) for donating the HPMC used, and Aria Pharmaceutical Co., for supplying diltiazem powder. REFERENCES
The following images related to this document are available:Photo images[pr09055f2.jpg] [pr09055f1.jpg] | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||

{kind=link}
{kind=link}