 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Tropical Journal of Pharmaceutical Research, Vol. 8, No. 6, December, 2009, pp. 531-537 Research Article Development and Validation of a RP-HPLC Method for the Simultaneous Determination of Rifampicin and a Flavonoid Glycoside - A Novel Bioavailability Enhancer of Rifampicin Bhusari S Sachin1, Vandhna Bhat1, Meenakshi Koul1, Subhash C Sharma1, Manoj K Tikoo1, Ashok K Tikoo1, Naresh K Satti2, Krishan A Suri2 and Rakesh K Johri1* Divisions of 1Pharmacology and 2Natural Products Chemistry, Indian Institute of Integrative Medicine, Jammu 180001, J & K, India. *Corresponding author: E-mail: rkjohri@iiim.res.in: Tel: +91-9419132014 Received: 30 April 2009 Code Number: pr09068 Abstract Purpose: To
develop and validate a sensitive HPLC method for the separation and simultaneous
estimation
of two ingredients in a composition comprising of rifampicin and a flavonoid
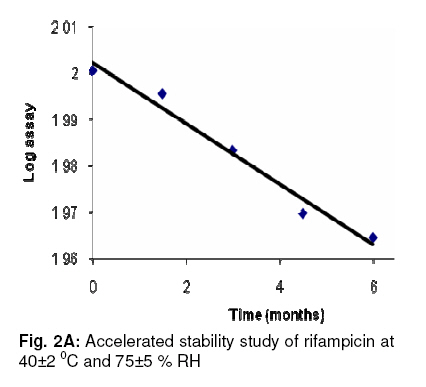
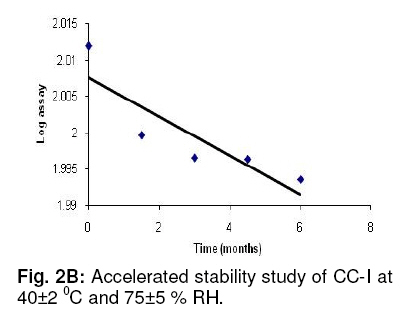
glycoside (an enhancer of oral bioavailability of rifampicin). Keywords: Flavonoid glycoside; RP-HPLC; Rifampicin; Stability studies; Validation INTRODUCTIONRecently, the use of natural products in combination with prescription drugs has attracted interest with regard to possible improvement in drug disposition profile since one of the major consequences of herb-drug interactions is altered drug bioavailability [1-3]. Consequently, herb-drug combinations are increasingly being examined as a useful strategy to enhance drug bioavailability [4]. In our earlier studies, a flavonoid glycoside isolated from Cuminum cyminum (cumin seed) and identified as 3¢, 5–dihydroxyflavone 7-O-b-D-galacturonide-4¢-O-b-D-glucopyranoside (CC-I) (Fig. 1), showed a pharmacokinetically meaningful interaction with rifampicin (RIF) , resulting in increased oral bioavailability of the drug. A significant increase in AUC (53 %) and Cmax (35 %) of RIF was evident when the drug was administered in combination with CC-I per orally in rats [5]. In view of the low/variable systemic bioavailability of orally administered RIF in a fixed drug combination [6], we intend to extend the pre-clinical studies of a composition consisting of RIF and this flavonoid glycoside (CC-I). Since incidences of possible drug interactions between concomitantly administered drugs are known to alter their pharmacokinetic profiles, we sought to develop and validate a RP-HPLC method for simultaneous estimation of both analytes (RIF and CC-I) in a single run. The present report deals with this method development in terms of specificity, recovery, linearity, accuracy and precision, as per the guidelines of International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) [7], and in the shortest possible time, for quality assurance and routine analysis of the blend. Accelerated and ambient stability studies are also included in this work. EXPERIMENTAL Reagents and chemicals RIF was purchased from Sigma (St. Louis, Mo, USA). All HPLC grade solvents and water were procured from Rankem, Mumbai, India. A 97 % pure CC-I (mol. formula, C27H28O17; MW, 624; melting point, 270 0C) sample was prepared as described earlier [5]. Based upon previous animal studies [5], the amounts of RIF and CC-I in the composition were adjusted to 450 mg and 50 mg, respectively. Instrumentation The HPLC system (Shimadzu, Japan) consisted of a diode array detector (SPD-M10AVP), solvent delivery module (LC-10ATVP), online degasser (DGU-14A), an auto-injector (SIL-10ADVP), flow channel system (FCV-14AH), system controller (SCL-10AVP), and a reversed-phase HPLC column (RP-18, 250 mm × 4.6 mm, 5 µm particle size, Sigma, USA). Data analysis was carried out using Class VP V6.12 SP2 software (Shimadzu, Japan). Stability studies were performed using a Climacell 222 stability chamber (BMT, Germany). A Cerius-2, 3D sketcher software was used for the determination of log P and hydration energy of CC-I. Chromatographic conditions The chromatographic elution was carried out in isocratic mode using a mobile phase consisting of acetonitrile and 50 mM phosphate buffer (pH 5.0) in a ratio of 60:40 v/v. The analysis was performed at 40 0C using a flow rate of 0.8 mL min-1 within a run time of 15 min. The 340 nm detection wavelength was found to be the same for both analytes. Preparation of reference solutions RIF (50 mg) was weighed accurately using a pre-calibrated weighing balance, transferred into a 50 mL volumetric flask, dissolved (by sonication) and diluted with acetonitrile to achieve 1 mg mL-1 strength (Stock I). CC-I (10 mg) was accurately weighed, transferred into a 10 mL volumetric flask, dissolved in HPLC grade water and diluted suitably to a final volume having a concentration of 1 mg mL-1 (Stock II). The flasks containing the reference solutions were covered with aluminium foil and sealed with paraffin film to avoid degradation and loss due to evaporation. Preparation of calibration, quality control and system suitability standards Stock I and II solutions were separately diluted serially with the mobile phase to obtain stock III (50 µg mL-1 of RIF) and stock IV (50 µg mL-1 of CC-I) solutions. Stock III and IV were mixed and then diluted with the mobile phase to obtain six calibration standards (0.1, 0.5, 1.0, 2.5, 5.0, and 10.0 µg mL-1), three quality control standards (0.1, 2.5, and 10.0 µg mL-1); and one system suitability standard (1.0 µg mL-1). Sample preparation from composition From three different batches of rifampicin-flavonoid glycoside mixtures, 18 samples (6 per batch) were selected and weighed. Empty shells were weighed to determine the average fill weight. After homogeneous mixing of the contents (using glass mortar and pestle), the quantities equivalent to 90 mg of RIF and 10 mg of CC-I (total weight: 100 mg capsule contents) were transferred to a 100 mL volumetric flask. Following the addition of a small quantity of the mobile phase (approximately 50 mL), the contents were intermittently vortexed and sonicated for 10 min to ensure complete dissolution of the contents (by visual inspection), and the volume was adjusted to mark with the mobile phase. The solution was mixed properly with shaking and filtered through a 0.45 µm pore filter into amber coloured glass vials. The filtrate was diluted suitably with the mobile phase to bring the concentrations of RIF and CC-I to fall within their respective calibration ranges (preliminary analysis data not shown). Method validation The method was validated in accordance with ICH guidelines. The parameters assessed were linearity, range, accuracy, precision, specificity, limit of quantitation and robustness. System suitability The system suitability test was performed using 9 replicate injections of system suitability standard before analysis of samples. The acceptance parameters of both RIF and CC-I were less than 0.5 and 1.5 % relative standard deviation (R.S.D.) for retention time and peak area, respectively, along with more than 3500 theoretical plates. The resolution (criterion > 3) was calculated using the formula: R = 1.18 [(t2-t1) / (W2 + W1)] , where t2 and t1 are the retention times of RIF and CC-I, respectively, and W2 and W1 are their respective peak widths at half height. Linearity and range Six calibration standards (0.1-10 µg mL-1 for RIF and 0.05-10 µg mL-1 for CC-I in combination) were utilised. The peak area vs concentration plots were subjected to linear least square regression analysis. The intra- and inter-day linearity values were established. Accuracy and precision Intra- and inter-day accuracy were established from quality control standards by evaluating nominal and mean measured concentrations of quality control standards which were compared and expressed as % difference (diff %). Diff % was calculated using the formula: Diff % = [(mean measured concentration - nominal concentration)/ nominal concentration] x 100. The intra and inter-day accuracy were also determined for the composition using spiked samples. The 100 mg contents of the composition (90 mg RIF + 10 mg CC-I) were spiked with a suitable combination of known stock solutions of RIF and CC-I to achieve a total content of 200 mg (180 mg RIF + 20 mg CC-I). The spiked contents were dissolved and the volume made up to 200 mL with mobile phase. The solution was then diluted serially with the mobile phase to achieve 3 solutions: solution A (0.90 mg RIF + 0.10 mg CC-I), solution B (1.80 mg RIF and 0.20 mg CC-I) and solution C (4.50 mg RIF + 0.50 mg CC-I). The intra- and inter-day precision (% RSD) - for both RIF as well CC-I - were established by analysing 9 replicates each of 3 quality control standards on day 1 and again on each of three consecutive days. Limit of quantitation (LOQ) The lowest concentration of calibration curve with acceptable accuracy and precision (% RSD < 15) was reported as LOQ for both analytes. The robustness of the method was evaluated by analysing quality control standards after multiple ratio adjustments in the mobile phase composition and pH of the aqueous buffer. The acetonitrile volume in the mobile phase was modified to between 55 and 65 %. The pH range selected for study was from 4.5 to 5.5. The acceptance criteria were less than 2 % variation in the final results after modification of the mobile phase composition and pH. Specificity The stability indicating criteria and specificity of the method were determined by using a mixture of stock I (reference solution of RIF) and stock II (reference solution of CC-I) in 1:1 (v/v) ratio (stock V). Stock V solution was divided into two parts: one part was stored at room temperature in the presence of light while the other part was stored protected from light. The analysis was performed on days 1, 3 and 6 after storage. The stability criteria were less than 2 % change in concentration after the specified storage. To ensure specificity of the method, stock V (containing both RIF and CC-I) was stressed under individual conditions for 24 h in: 1.0 N HCl, 1.0 N NaOH, 30% w/v H2O2, and 50 mM Tris buffer pH 8.0. After 24 h, samples were analysed for RIF and CC-I. The peak purity was considered as a parameter and evaluated using photo-diode array detector and peak purity software. The upslope, apex and downslope portions of the peak were compared by superimposing the respective chromatograms. The peak was considered pure only when identical spectra were found at every portion of the peak. RESULTS The following HPLC conditions showed best resolution of RIF and CC-I in terms of retention time, peak characteristics and total run time: (a) mobile phase - acetonitrile : KH2PO4 buffer (pH 5, 50 mM) 60:40 (v/v); (b) flow rate - 0.8 mL min-1; (c) column head pressure – 808 - 838 psi; (d) column - RP-18 (250 x 4.6 mm, 5 µm); and (e) column oven temperature, 40 0C. The respective retention time of RIF and CC-I was 4.779 and 3.072 min under the above HPLC conditions. System suitability test showed that critical parameters such as retention time, area and number of theoretical plates met the acceptance criteria on all the experimental days (Table 1). Table 1:
System suitability test
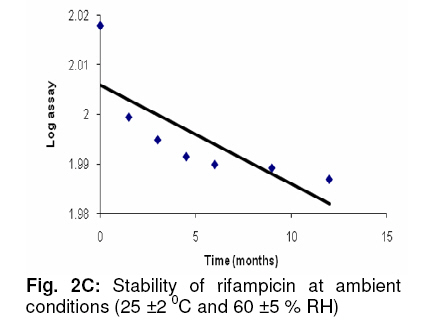
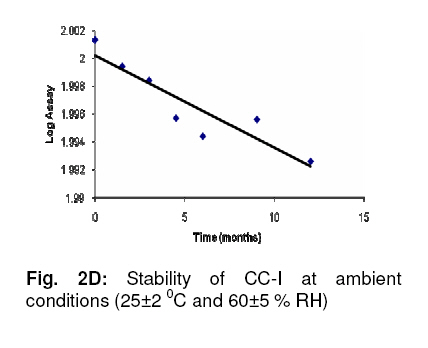
Adequate resolution of > 6 between RIF and CC-I peaks was observed. The linearity and range of calibration curves were assessed by constructing standard curves for RIF (0.10-10.00 µg mL-1) and CC-I (0.05-10.00 µg mL-1). Analysis of three independent sets showed good correlation between concentration and resulting peak area for both analytes (RIF: slope, 0.48, y-intercept, 2.26, and r2, >0.999; CC-I: slope, 0.00046, y-intercept, 0.24351, and r2, >0.999). The accuracy and precision were established by using 9 replicates each of 3 different quality control standards. The intra- and inter-day accuracy (diff. %) for RIF was in the range of −3.34 to +3.28 and −4.87 to +2.88 respectively, while for CC-I, it was from −4.59 to +3.42 and −3.62 to +3.50, respectively. The intra- and inter-day precision (% RSD) for RIF was in the range of 1.08 to 2.61 and 1.62 to 2.77, respectively, while for CC-I, it was 1.22 to 2.66 and 1.14 to 2.98, respectively. RIF + CC-I mixture was subjected to batch-wise accuracy measurement after spiking 50 % of the known amounts of analytes. The intra- and inter-day accuracy (diff. %) of RIF was −2.932 to +2.78 and −3.92 to +2.17, respectively, and for CC-I, it was −3.61 to +3.60, and −5.01 to +2.34, respectively. Based on the acceptable accuracy and precision, the LOQ of RIF and CC-I were 0.10 and 0.05 µg mL-1, respectively. Minor modifications of mobile phase composition and pH of buffer did not significantly alter the performance of the method in terms of retention time, resolution, peak characteristics and theoretical plates. CC-I solution was stable at all the storage conditions for at least 6 days. The reference solution of RIF, after storage at room temperature for 24 h in the presence of light, showed 9.5 % degradation (established on the basis of peak purity). The stability studies were performed at ambient and accelerated conditions (in compliance with regulatory requirements) of 25 ± 2 0C and 60 ± 5 % RH, and 40 ± 2 0C and 75 ± 5 % RH, respectively. The results showed a reduction of RIF content from 100.2 to 92.2 % and followed first order kinetics (Fig 2A). CC-I content declined from 102.8 to 98.5 % (Fig 2B). A 12 month study showed the average shelf life of 22.89 months (on the basis of RIF). DISCUSSION CC-I, a flavonoid glycoside, is a highly polar compound. It is expected to be poorly retained on reverse phase HPLC column, hence the smaller retention time. RIF, on the other hand, is relatively non-polar and showed stronger retention. Simultaneous analysis of the two analytes, which are chemically different, in a single run under similar chromatographic conditions was a difficult task. Ensuring the quality of the composition in terms of RIF and CC-I assay and minimising the analysis time for the assays were the main objectives of the study. Therefore, a method has been developed for the analysis of both ingredients in a mixture simultaneously under similar chromatographic conditions and in the shortest possible time. The method development involved optimisation of the buffer - its strength and pH. Several stationary and mobile phases were tried. A range of columns of different materials were tested, along with different mobile phase compositions. In preliminary studies, various buffers from lower to medium strength were combined with methanol or acetonitrile (data not shown). Finally, method parameters such as mobile phase composition, its pH, flow rate, etc, were optimised on the basis of peak characteristics and run time. The method was validated for its performance in terms of linearity, range, accuracy, precision, LOQ, robustness and specificity for both RIF as well as CC-I. The ICH guidelines were followed for validating the method. To ensure the validity of a system and an analytical method, system suitability test was performed. Adequate resolution between RIF and CC-I peaks showed the efficiency of the method to identify and quantify each analyte at the same time with no interference. The method was found to be robust as minor intentional changes in the method parameters did not alter method performance. Based on the stability data of RIF solution, a fresh RIF solution needs to be prepared each time before analysis. The validated method was utilized successfully for determining the stability of the composition at ambient conditions. CONCLUSION In the present investigation, a simple, accurate, precise and robust RP-HPLC method has been developed for the determination of both ingredients (RIF and CC-I) in a composition. The method has potential application in quality evaluation and prospective clinical investigations. ACKNOWLEDGEMENT This work was supported by an extramural grant (Senior Research Fellowship) of Council of Scientific Research, New Delhi, India. REFERENCES
© Pharmacotherapy Group, Faculty of Pharmacy, University of Benin, Benin City, 300001 Nigeria. The following images related to this document are available:Photo images[pr09068f2d.jpg] [pr09068f1.jpg] [pr09068f2b.jpg] [pr09068f2c.jpg] [pr09068f2a.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}