 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Tropical Journal of Pharmaceutical Research, Vol. 9, No. 1, 2010, pp. 51-58 Research Article Preparation, Characterisation and In vivo Evaluation of Bis-demethoxy Curcumin Analogue (BDMCA) Nanoparticles CA Anuradha and Jithan Aukunuru* Vaagdevi College of Pharmacy, Ramnagar, Hanamkonda, AP, India-506001 *Corresponding author: E-mail: aukunjv@gmail.com; Tel: 0091-870-2455111 Received: 17 June 2009 Code Number: pr10007 AbstractPurpose: To fabricate biodegradable nanoparticle formulation
of bis-demethoxy curcumin analogue (BDMCA), a novel curcumin analogue, and
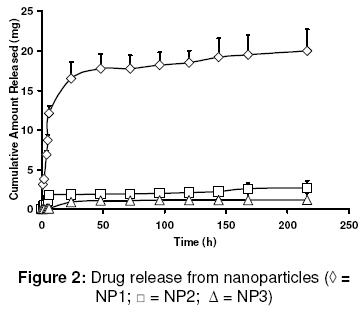
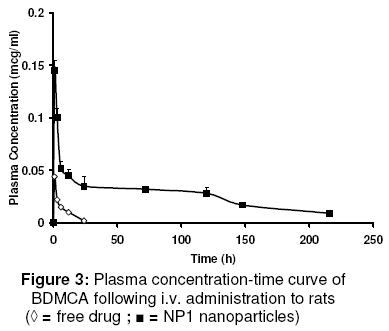
evaluate its in vitro and in vivo characteristics. Keywords: Curcumin analogue; Nanoparticles; pharmacokinetics; Pharmacodynamics; Hepatoprotective activity INTRODUCTION Curcumin has demonstrated several pharmacological activities and therapeutic applications in a variety of disease states [1]. However, it shows poor systemic delivery. Data from several studies indicate that its poor solubility, very low GIT dissolution rate, low absorption, and extensive systemic metabolism are the reasons for its delivery problems and lack of clinical success [2]. Several formulations as well as drug discovery strategies have been investigated to overcome these delivery issues. In order to improve the delivery of curcumin, phosphatidylcholine formulations, lipid-complexes, solid-dispersions, prodrugs, microspheres, analogues, derivatives, and nanoscale formulations are among those that have been investigated [3-7]. The structures of curcumin and bisdemethoxy curcumin analogue (BDMCA), a novel curcumin analogue which is the subject of this study, are shown in Fig. 1. Several analogues with improved pharmaceutical properties, as well as therapeutic and pharmacological activities have been synthesised [1]. Identification of newer curcumin analogues with improved activities is of significant preclinical and clinical interest. The hepatoprotective activity of BDMCA has previously been reported [8]. In chronic diseases, such as cirrhosis and associated liver disorders for which hepatoprotection is required, drugs are usually given daily by the oral route. However, such a practice is frequently associated with peak systemic drug levels which are either toxic or subtherapeutic. A suitable sustained release system would prolong drug release at the site of action and minimise fluctuations in drug levels, thus resulting in effective therapy. Additionally, targeting of drugs to the cells involved in a disease can increase therapeutic effectiveness several-fold. Therefore, an attempt was made in this study to develop a biodegradable nanoparticulate formulation which not only can sustain drug release but also lead to enhanced intracellular levels of the drug in liver cells, such as Kupffer cells, which are not only exposed to systemic circulation and but are also phagocytic in nature [9]. Sinusoidal endothelial cells, which also exposed to systemic circulation, can actively take up particles by endocytosis [10]. Both of these cell types are known to be involved in the pathology of cirrhosis and associated liver disorders [11]. The first objective of this study was to fabricate and characterise nanoparticles of BDMCA using a double-emulsion solvent evaporation technique and with polycaprolactone as the polymeric material. To address the drug delivery shortcomings of BDMCA molecule and thereby alter its pharmacokinetic and pharmacodynamic properties, release of the drug through polymeric material is of immense importance. Thus, the second objective of this work was to evaluate the pharmacokinetics and hepatoprotective activity of the drug following intravenous administration of the developed formulation. Advantages of delivery systems such as this have previously been reported [12,13]. EXPERIMENTAL Material All the chemicals and reagents used were purchased from Finar Chemicals and Merck Specialities, both in New Delhi, India. Polycaprolactone was purchased from Sigma-Aldrich Taufkirchen, Germany while polyvinylalcohol was obtained from Qualikens Fine Chemicals, New Delhi, India. Male albino Wistar rats, weighing 150 - 250 g, and purchased from Mahaveera Enterprises, Hyderabad, India, were used in the in vivo investigation. Bis-demethoxy curcumin analogue (BDMCA) was synthesised and purified in our laboratory as previously described [7,14]. Its purity was further checked with HPLC using methanol:water at a ratio of 80:20 as a mobile phase at a detection wavelength of 230 nm. Preparation of BDMCA nanoparticles BDMCA nanoparticles were prepared using a double emulsion-solvent evaporation technique. Three different formulations NP1, NP2, NP3 containing drug:polymer in the ratio of 1:1.5, 1:2 and 1:4, respectively, were prepared. Briefly, dichloromethane (10ml) containing the drug (100 mg) and the polymer dissolved in it, was taken as the organic phase. The aqueous phase (5 ml), containing polyvinyl alcohol in water (to form a 3 %w/v solution), was added to the organic phase and sonicated using an ultrasonicator (Homogenizer 150 VT, M/S Biologics, Inc, USA) for 10 min to form a w/o emulsion. The w/o emulsion was subsequently added to 30 ml of 3 % w/v polyvinylalcohol solution and sonicated for 5 min to form a w/o/w emulsion. The emulsion was stirred on a magnetic stirrer until complete evaporation of dichloromethane occurred, leaving a nanoparticle suspension. The suspension was centrifuged at 10,000 rpm for 30 min, resulting in the formation of a pellet at the bottom of the tube. This pellet was re-suspended in phosphate buffered saline (PBS) to form a nanoparticle suspension which was used for further evaluation. Aseptic technique was used in the preparation of sterile nanoparticle formulations used in the animal experiments. In this case, prior to the preparation in a laminar flow hood, all the excipients and the containers used were sterilized in a formalin chamber. Characterization of BDMCA nanoparticles The nanoparticle formulations were evaluated for particle size and charge, drug encapsulation, drug-excipient interaction and in vitro drug release. Determination of particle size and charge Values of effective hydrodynamic diameter in dilute dispersions were obtained by dynamic light scattering measurements using a zeta sizer (ZetaPlus zeta potential analyzer, Brookhaven Instruments Corp, Holtsville, NY, USA) equipped with a laser source at a fixed angle of 90º. Prior to analysis, the solutions were filtered through a filter (pore size ≈ 2 mm) to remove dust. Evaluation of encapsulation efficiency The amount of drug encapsulated in the particles was determined using the dialysis method. The drug that diffused into the receptor compartment across the dialysis membrane (MW: 25,000) was assayed at 425 nm wavelength using a UV-Vis spectrophotometer (Elico SL 164) and the cumulative amount released was calculated. The volume of the receiver compartment was 1 L. Drug release was monitored until no more drug was released for approximately 30 min. In vitro release experiments The drug (free or encapsulated in nanoparticles) was placed in the donor compartment of a dialysis permeability cell containing 100 ml of phosphate buffered saline (PBS) with a dialysis membrane (MW: 25,000) separating the donor and receptor compartments. At predetermined intervals, the dialysate was sampled and the amount of BDMCA was determined spectrophotometrically at 425 nm. Drug release was monitored until no more drug was released for 30 min. Studies on drug-polymer interaction Drug-polymer interaction was investigated using a Fourier transform infrared (FTIR) spectroscopy. The FTIR spectra of the drug, polymer, blank nanoparticles, and drug-loaded nanoparticles were taken with a Thermo Nicolet Nexus 670 spectrometer using KBr pellets. Pharmacokinetic studies The pharmacokinetics of the formulation was performed in two groups of male Wistar rats of six rats each – one group for the nanoparticles and the other for the reference drug. All the animal experiments were conducted according to guidelines of the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Chennai, India [15] and the study protocol was approved by Institutional Animal Ethical Committee of Vaagdevi College of Pharmacy, Warangal, India (ref no. 1047/ac/07/CPCSEA). The rats were acclimatised for 10 days prior to the commencement of the experiment (12/12 hour dark/light cycle at a temperature of 20 0C and relative humidity of 60%) and fed on standard diet. These conditions were maintained throughout duration of the experiment. Nanoparticle formulation, NP1, was used in the in vivo studies because it had the smallest particle size, high encapsulation efficiency and fastest in vitro drug release. An amount (10 mg) of the nanoparticules (equivalent to 7 mg of the drug), dispersed in 1ml of normal saline, was injected intravenously into the rats via the tail vein. For the reference, 7 mg of the drug was dissolved in 1 ml PEG 400 and administered i.v to the rats via their tails. Blood samples were collected at 0.5, 1, 2, 3, 6, 12, 24 h and then 3, 6 and 9 days. Plasma BDMCA was estimated using HPLC (Cyberlab, USA). Prior to this, HPLC standard curve for the drug in plasma was generated. The UV detection wavelength was 230 nm. The mobile phase consisted of methanol:water at a ratio of 80:20. The following pharmacokinetic parameters (for both reference and nanoparticle administrations) were determined using WinNonlin pharmacokinetic data analysis software: elimination rate constant (ke), volume of distribution (Vd), elimination half-life (t1/2), clearance (CL), and area under curve (AUC). Evaluation of hepatoprotective activity Hepatoprotective activity was used as a measure of pharmacodynamic activity. It was determined for the reference (pure BDMCA) as well as the nanoparticles. Male albino Wistar rats, weighing 150-200 g, were divided into 6 different groups. Group I was normal control; Group II, which served as toxic control, received a mixture of CCl4 and olive oil (25:75) at a dose of 0.7ml/kg by the intraperitonial route on the 3rd, 6th and 9th day. Group III received 3.5 mg BDMCA dissolved in PEG 600 (0.5 ml) by intravenous route twice daily, giving a total daily dose of 7 mg. Group IV received 10 mg of BDMCA nanoparticle formulation (NP1), equivalent to 7 mg of BDMCA, once at the beginning of the experiment via the i.v. route. Group V animals were injected i.v. with blank nanoparticles and Group VI animals served as control (for PEG 600) and were injected 1 ml of PEG 600 twice a day. Hepatoprotective activity was quantified by assessing serum glutamic oxaloacetic transaminase (SGOT) and serum glutamine pyruvic transaminase (SGPT) levels as well as histological features following the procedure reported previously [15,16]. The body weights of the rats were also monitored. Statistical analysis The data were presented as mean ± standard deviation (S.D.) and were analysed by 2-tailed Students t-test using MiniTAB software (Minitab Inc. State College, PA, USA). P < 0.05 was used in determining statistically significant difference. RESULTSThe purity of BDMCA, as assessed by HPLC, was 99.7 %. As Table 1 shows, drug/polymer ratio did not exert any significant influence (p < 0.05) on the particle size of the formulations. However, at a drug/polymer ratio of 1:1.5 (formulation NP1), encapsulation efficiency was high (70 %), polydispersity was least and the particles carried a significant negative charge. Consequently, this formulation was used in subsequent studies. Drug-polymer interaction The FTIR spectra of BDMCA, BDMCA nanoparticles, blank nanoparticles and pure poly(caprolactone) (not shown) indicate that there was no interaction between the drug and the polymer. Furthermore, the characteristics of the polymer were not altered during the formulation process. In vitro drug release characteristics In vitro release data, shown in Fig 2, indicate that all the three formulations sustained drug release for over a period of 9 days. Drug release from NP1 was several fold-higher than from NP2 and NP3. This could be due to stronger binding of the drug to the polymer in NP2 and NP3 which had lower drug concentrations than NP1. Higuchi plot of the data (not shown) indicate that all the formulations exhibited burst release on the first day and thereafter, drug release followed Higuchi pattern up to the 9th day of the study. This suggests diffusion-mediated release after an initial burst release. Pharmacokinetics of the formulations Extraction efficiency of the method used was 85 %. The retention time of the drug was 8 min and the minimum detection level was 0.1 mg/ml. Plasma profiles of the drug following i.v. administration of the BDMCA nanoparticles and the free drug (reference) are plotted in Fig. 3. BDMCA nanoparticles resulted in a higher and prolonged drug levels, which refleced as increased area under the curve. T1/2b, Vd, AUC, and CL for NP1 formulation were 150 ± 20 h, 1.33 ± 0.2 L, 11.2 ± 0.2 mg.hr/ml, and 1.2 ± 0.4 ml/min, respectively, while the corresponding values for the free drug were 3.46 ± 1.50 h, 0.08 ± 0.04 L, 0.25 ± 0.15 mg.hr/ml, and 6.4 ± 1.2 ml/min, respectively. Thus, the half-life (t1/2), volume of distribution (Vd), and the area under the curve (AUC) of NP1 formulation were higher (P < 0.05) than the corresponding values for the free drug, while clearance (CL) was lower than for the free drug. Hepatoprotective activity Table 2 shows the hepatoprotective data for NP1 and controls. The results show a significant decrease in the levels of SGOT and SGPT in the rats that received the nanoparticles when compared with the levels of the enzymes in the CCl4–treated rats. The PEG control as well as blank nano-particles did not exert any change on SGOT and SGPT levels suggesting that these materials were non-toxic to the rat liver. The histopathological data tallied with the biochemical data as there was significant necrosis of the liver cells including the parenchyma in CCl4–treated rats while in the other groups, the histological assessment revealed normal liver. While administration of the free drug to CCl4-treated rats lowered both SGOT and SGPT levels significantly, it did not, however, achieve complete reduction to normal levels. However, both BDMCS preparations restored the histological status of the liver of the CCl4-treated rats to normal. Table 1: Some properties of BDMCA nanoparticles
Table 2: Hepatoprotective activity of the preparations in male Wistar rats
SGOT = serum glutamic oxaloacetic transaminase; SGPT = serum glutamic pyruvic transaminase Furthermore, the body weights of the rats which fell significantly after CCl4 treatment, were restored to normal following administration of both BDMCA preparations. DISCUSSION One of the problems with particulate drug carriers including nanomaterials is the possibility that they may be engulfed by the mononuclear phagocytic system of the liver and the spleen [9,10]. However, such an occurrence can be an added advantage when treating liver diseases such as tumour metastasis, fibrosis, cirrhosis or hepatitis, where phagocytic cells are likely to be involved. The present study is premised on such an approach since BDMCA, a curcumin analogue, has been found to be useful in liver diseases, such as cirrhosis, which involves several liver cells. Onset of fibrosis/cirrhosis starts with the initiation of biochemical cascade in Kupffer cells as well as sinusoidal endothelial cells, both of which can take up nanoparticles at a significant rate [11]. Thus, drugs when encapsulated in the nanoparticles and administered via the i.v. route, can lead to very high intracellular levels in liver cells. For instance, the delivery of flavopiridol using a novel liposomal formulation resulted in better pharmacokinetic properties than the solution form [13]. Similarly, paclitaxel nanoparticles not only showed effectiveness in in vitro cell culture models but also increased sustainable therapeutic efficacy in an in vivo animal model [12]. The paclitaxel was encapsulated in a vitamin E TPGS-emulsified PLGA nanoparticles, and this system resulted in a higher and prolonged level above the effective concentration in vivo, as reflected by increased area under the curve. BDMCA nanoparticles were successfully prepared using the protocol described in this study. NP1 was the most suitable of the three nanoparticle formulations prepared but it could be improved on and even tailored to meet various needs. These nanoparticles sustained the release of the drug both in vitro as well as in vivo. The hepatoprotective studies demonstrated effective reversal of hepatotoxicity by the nanoparticles but the reversal was not 100 %, probably due to the administration of insufficient doses. Thus, a higher dose may be required to achieve 100 % reversal of elevated SGOT and SGPT levels to normal. The rationale for nanoparticle entrapment of drugs include enhancement of drug delivery to, or uptake by, target cells, and/or a reduction in the toxicity of the free drug at non-target organs. Both of these situations will result in increase in therapeutic index, the margin between the doses resulting in therapeutic efficacy (e.g., tumour cell death) and toxicity to other organ systems. Based on the findings of this study, BDMCA appeared to have shown potentials for meeting these desirable objectives. CONCLUSION Nanoparticles of BDMCA can be conveniently prepared by a solvent evaporation technique using polycaprolactone as a biodegradable polymer. The particles sustained the drug release both in vivo and in vitro. The nanoparticles improved the pharmacokinetic properties of the drug. BDMCA nanoparticles were more effective than the free drug in achieving reversal of biochemical changes induced by the administration of CCl4. The nanoparticles can be further developed to enhance delivery of BDMCA to the liver of rats. ACKNOWLEDGEMENT The authors would like to acknowledge the management of Vaagdevi College of Pharmacy, Warangal, for providing facilities for this work. One of the authors, Dr Jithan Aukunuru, is grateful to the Department of Science and Technology, India, for granting financial assistance that enabled him to participate in this study. This work was largely funded with a SERC-DST Young Investigator project grant to Dr. Jithan Aukunuru. REFERENCES
© Pharmacotherapy Group, Faculty of Pharmacy, University of Benin, Benin City, 300001 Nigeria. The following images related to this document are available:Photo images[pr10007f1.jpg] [pr10007f2.jpg] [pr10007f3.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}