 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
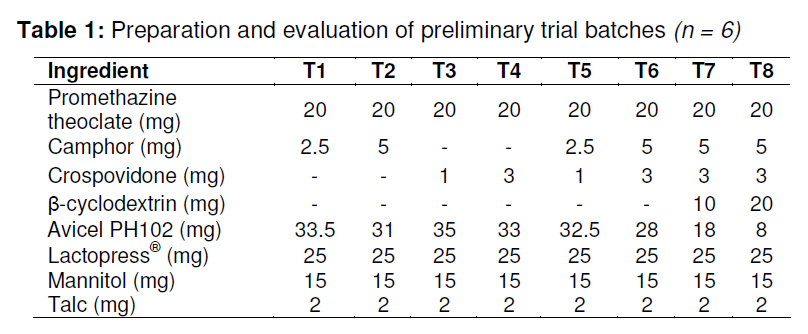
Tropical Journal of Pharmaceutical Research, Vol. 9, No. 5, September-October, 2010, pp. 489-497 Research Article Formulation of Fast-Dissolving Tablets of Promethazine Theoclate Shailesh Sharma*, Neelam Sharma and Ghanshyam Das GuptaDosage Form Design Laboratory, Pharmaceutics Research Division, Department of Pharmaceutics, ASBASJSM College of Pharmacy, BELA (Ropar) Punjab 140111, India *Corresponding author: Email: shailesh.bela@gmail.com; Tel: +91-9888 775589; Fax: +91-1881-263108 Received: 24 February 2010 Revised accepted: 30 August 2010 Code Number: pr10059AbstractPurpose: To optimize and formulate promethazine theoclate fast-dissolving
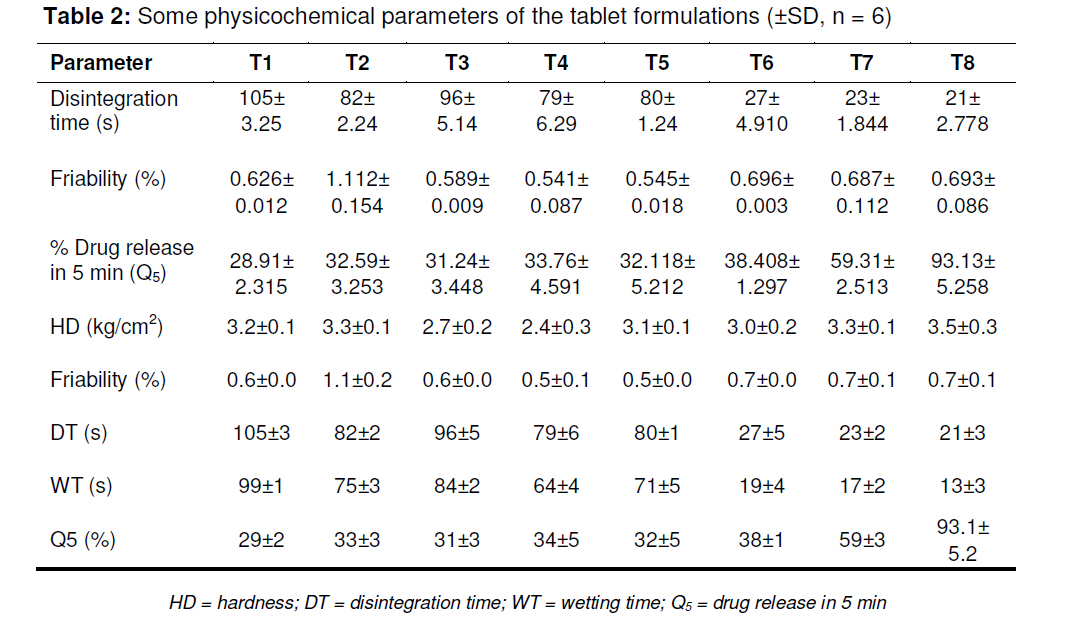
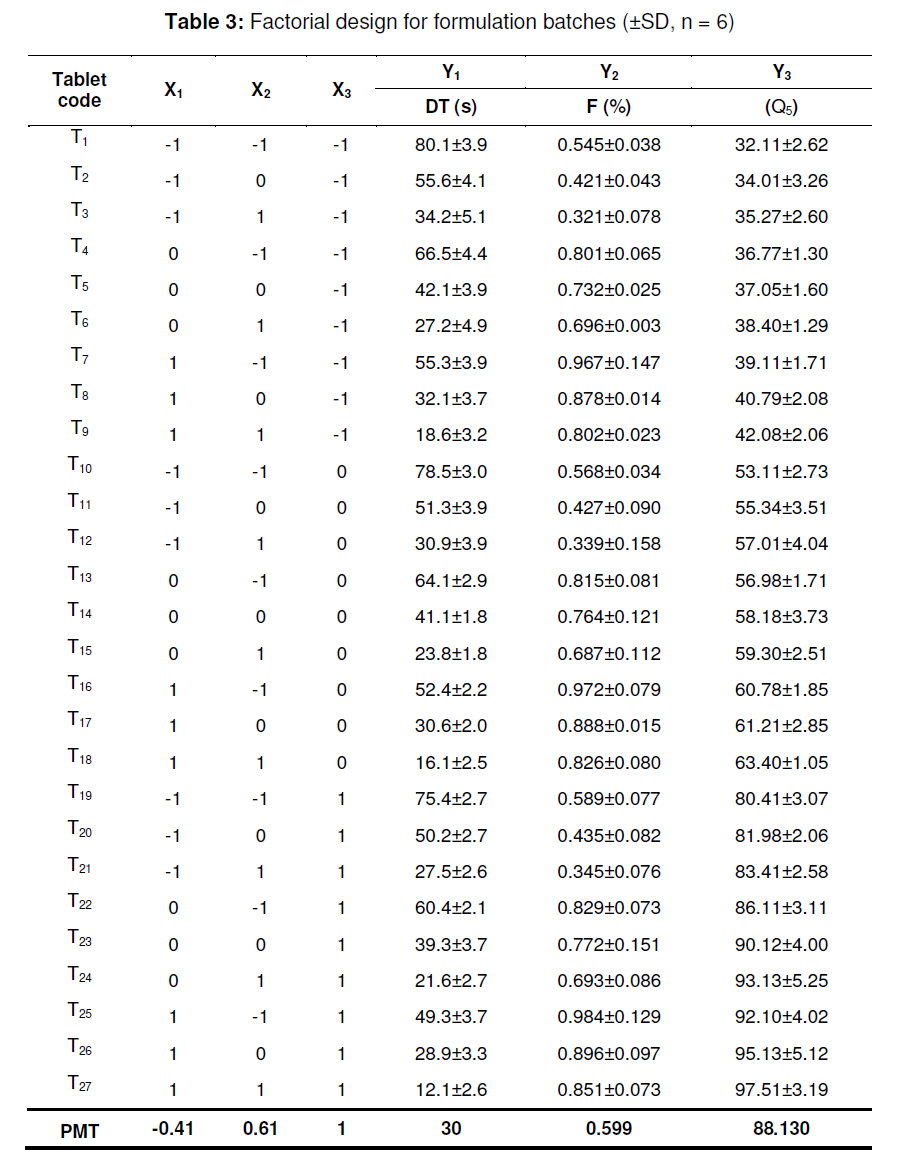
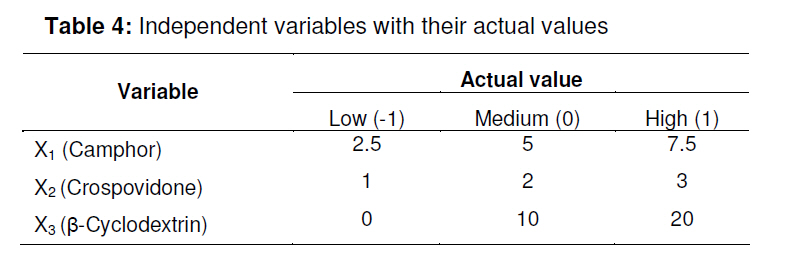
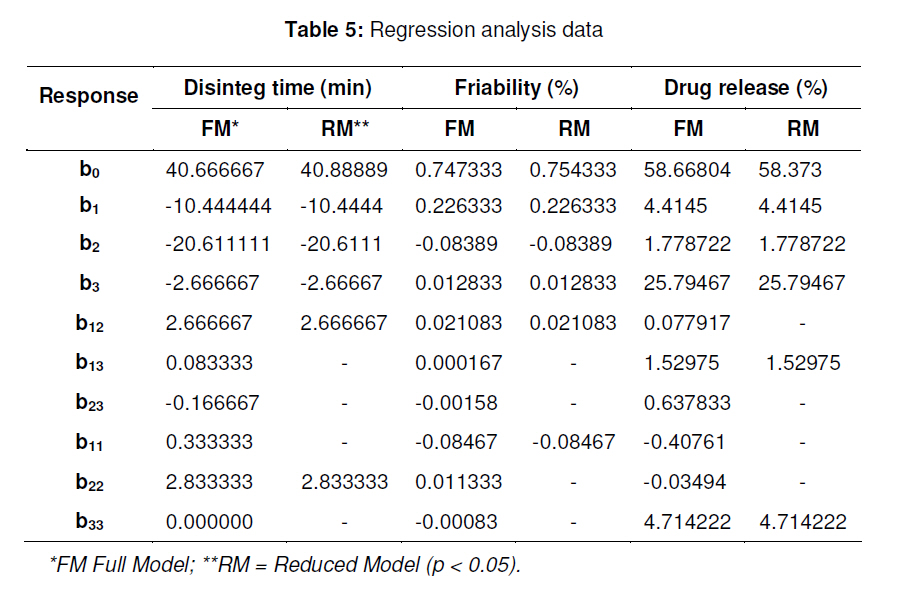
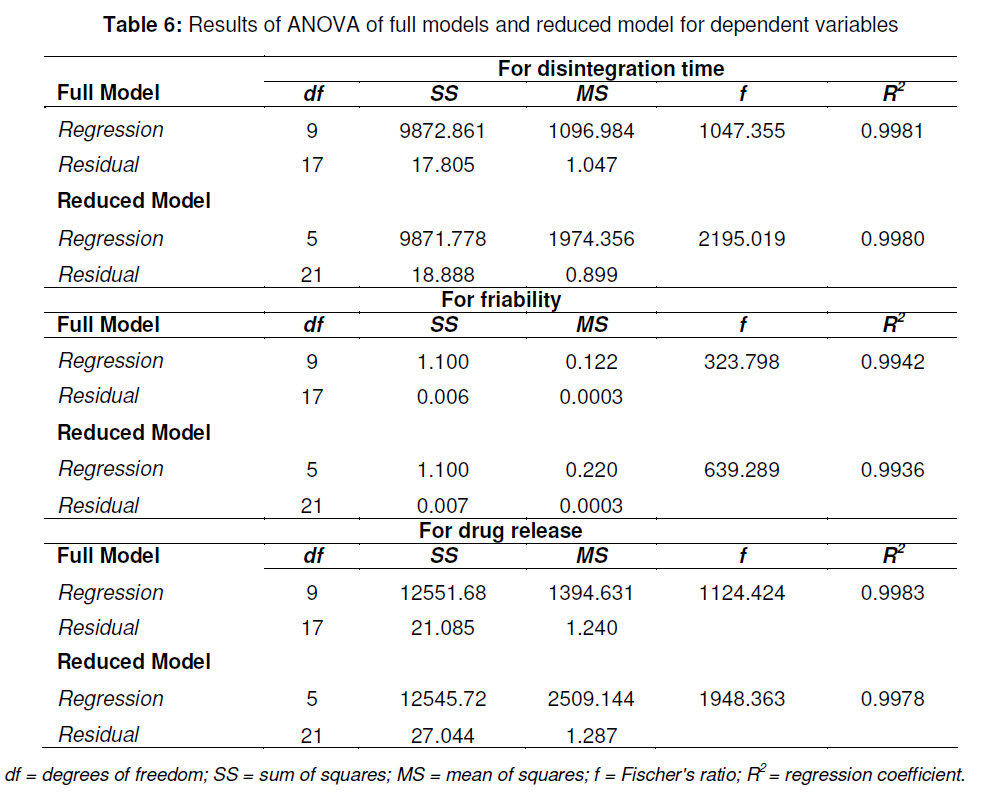
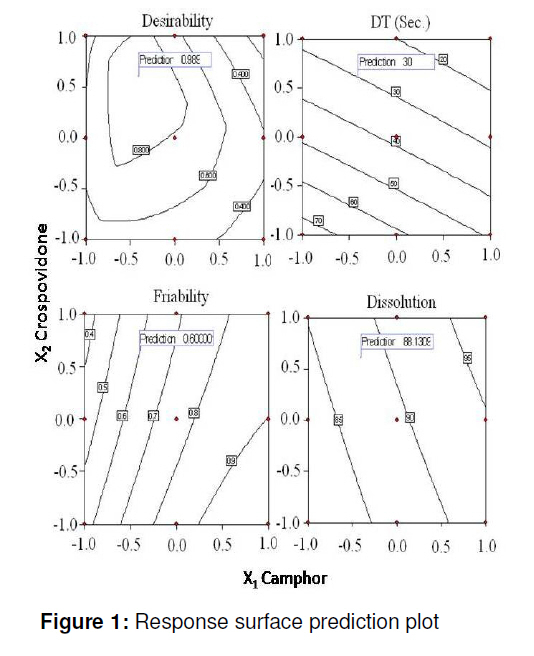
tablets that offer a suitable approach to the treatment of nausea and vomiting. Keywords: Fast-dissolving tablet, 33 Factorial design, Promethazine theoclate, Optmization studies. INTRODUCTION Retention of an administered antiemetic oral dose and its subsequent absorption during therapy is critically affected by recurrent emesis, a process coordinated by the vomiting centre in the lateral reticular formation of the medulla receiving inputs from the chemoreceptor trigger zone and other neural sites [1]. Vomiting induced by physiological processes such as impaired gastric emptying and other gastric disturbances will also affect drug retention and absorption [2]. Retention of oral dose is, therefore, a prerequisite for absorption to prevent emesis. For drug with low bioavailability, partial drug loss by emesis will result in therapeutic failure. One such antiemetic drug, promethazine theoclate, after oral dosing, undergoes extensive gastric and first pass effect. This results in low bioavailability which, therefore, will not minimize the rate of vomiting [3]. A fast dissolving system can be defined as a dosage form for oral administration, which when placed in the mouth, rapidly disperses or dissolves and can be swallowed in the form of liquid [4]. Fast-dissolving tablets of promethazine theoclate are designed for rapid and complete absorption in the gastrointestinal tract in order to achieve therapeutic success. Fast-dissolving formulations are popular because they are easy to administer and lead to improved patient compliance. Paediatric and geriatric patients have difficulty swallowing (dysphasia) conventional dosage forms [5]. Fast-dissolving drug delivery systems may offer a solution to this problem. This dosage form dissolves or disintegrates in the oral cavity within a minute without the need of water or chewing [6]. The basic approach to the development of fast dissolving tablets (FDT) is the use of superdisintegrants. Another approach is maximizing the pore structure of the tablets. Freeze-drying [7,8] and vacuum-drying [9,10] techniques have been tried by researchers to maximize the pore structure of the tablet matrix. Freeze drying is cumbersome and yields a fragile and hygroscopic product. Therefore, the vacuum-drying technique was adopted in the present study. Full factorial experimental design is one of the best tools for studying the effect of different variables on the quality determinant parameters of any formulation. Multiple regression analysis of results gives an equation that adequately describes the influence of the independent formulation variables on the selected responses [11]. The objective of the present work was to develop fast dissolving tablets of promethazine theoclate based on a small number of experimental runs [12]. Use of a 33 factorial design was attempted to generate an optimized region in the contour plots where the combination of β-cyclodextrin (solubility enhancer), camphor (pore forming agent) and crospovidone (superdisintegrant) could provide hard and rapid disintegrating tablets which can release the drug maximally within 5 min. EXPERIMENTAL Materials Promethazine theoclate and crospovidone were gifts from Mehta Pharmaceuticals, Mumbai, India and BASF Chemicals, Mount Olive, NJ, USA, respectively. β-cyclodextrin, Lactopress®(lactose anhydrate) and microcrystalline cellulose (Avice PH102) were also obtained as gifts from Signet Chemicals, Mumbai, India. Camphor, mannitol, talc and magnesium stearate were purchased from Ranbaxy Chemicals, India). All other chemicals used were of analytical grade. 33response surface model factorial design The traditional approach to developing a formulation is to change one variable at a time. By this method it is difficult to develop an optimized formulation, as the method reveals nothing about the interactions among the variables. Hence, a response surface design model with 3 factors, 3 levels, and 27 runs was selected for the optimization study. A 33 randomized full factorial design was used in the present study. In this design, 3 formulation independent factors are evaluated, each at 3 levels (low, medium and high), and experimental trials are performed at all 27 possible combinations. The amount of subliming agent, camphor (X1), the amount of superdisintegrant, crospovidone (X2), and the amount of solubility enhancer, βcyclodextrin (X3) were selected as independent variables. The disintegration time (DT), percentage friability (% F) and drug release in five minute (Q5) were selected as dependent variables. After application of full factorial design and with the aid of produced polynomial terms, the amount of three formulation variables was optimized. The optimized amount of the camphor, crospovidone and β-cyclodextrin were incorporated in the tablet which was used as the check point of the regression analysis model. The polynomial equation generated by this experimental design (using Design Expert 7.1.6 software, State Ease Inc) is as follows: Y= b0 + b1X1 + b2X2+ b3X3+ b12X1 X2+ b13X1 X3+ b23X2 X3 + b11X1 X1 + b22X2 X2 + b33X3 X3............. (1) where Y is the dependent variable; b0 is the intercept; b1 to b33 are the regression coefficients; and X1, X2 and X3 are the independent formulation variables [12]. Preparation of promethazine theoclate tabletsThe composition of the preliminary and factorial design batches are shown in Tables 1 and 2, respectively. All the raw materials were passed through a screen of 450 µm aperture size prior to mixing. Promethazine theoclate, camphor, crospovidone, microcrystalline cellulose, mannitol and lactose were mixed dry using a glass mortar and pestle. The blends were lubricated with 2 %w/w each of talc and magnesium stearate. The blends were compressed in a singlepunch tablet machine (Cadmach, Ahmedabad, India) to approx. 100 mg convex-faced tablets with a diameter of 5 mm. The tablets were dried for 6 h under vacuum (30 Kpa) at 50 oC to render the tablets porous by sublimation of the camphor. Evaluation of tablet propertiesThe crushing strength of the tablets was measured using a Monsanto hardness tester while tablet friability was assessed with a Roche friabilator. Twenty pre-weighed tablets were rotated at 25 rpm for 4 min and then reweighed after removal of fines (using 250 µm aperture screen), and the weight loss (%) was calculated. The wetting time of the tablets was determined using a simple procedure [13]. Five circular pieces of tissue paper (10 cm diameter, 0.45 µm pore size, Hi-media Corp) were placed in a 10 cm diameter Petri dish. Ten millilitres of water containing a water-soluble dye, eosin (0.01 %), was added to the Petri dish. A tablet was carefully placed on the surface of the tissue paper. The time required for water to reach the upper surface of the tablets was noted as the wetting time [14]. A modified method was used to determine the disintegration time and dissolution profile of the tablets to simulate conditions in the oral cavity. To assess disintegration time, 6 ml of Sorenson’s buffer (pH 6.8) at 37 ± 0.5 ° C was placed inside a 10 ml cylindrical glass vessel in such a way that 2 ml of the media was below the sieve and 4 ml above the sieve. One tablet was placed on the sieve and the whole assembly was then mounted on a high precision water bath shaker (Narang Scientific Works, India). The time taken for all the particles to pass through the sieve was noted as the disintegration time of the tablet. Six tablets, selected randomly from each batch, were tested and the mean value was calculated [15]. To determine dissolution profile, the apparatus employed for disintegration test was also used. Samples (1 ml) were withdrawn at different time intervals and replaced with fresh medium. The samples were filtered, diluted with Sorenson’s buffer (pH 6.8) and analyzed spectrophotmetrically at 250 nm. Data analysisA response surface model factorial design with 3 independent formulation variables at 3 different levels were used to study the effects on dependent variables. All the batches of fast dissolving tablets were statistically (p < 0.05) evaluated with regard to disintegration time, friability and drug release by using software Design Expert version 7.1.6 (State Ease Inc.). RESULTSThe results, shown in Table 2, indicate that concentration-dependent disintegration was observed in all batches prepared using camphor as a subliming agent (T1 and T2) and crospovidone as superdisintegrant (T3 and T4). Tablets containing combinations of subliming agent and superdisintegrant (T5 and T6) showed the least disintegration time. The effect of β-cyclodextrin was obvious; as affecting the formulation and optimize the fast the amount of β-cyclodextrin increased (T6, dissolving tablet for a disintegration time of T7 and T8), the release rate of the drug also 30 s, friability of < 0.6 %, and drug release of increased 2-to 3-fold. up to 90 %. The transformed values for all the batches along with their results are A factorial design was employed in order to systematically investigate the factors affecting the formulation and optimize the fast dissolving tablet for a disintegration time of 30 s, friability of < 0.6 %, and drug release of up to 90 %. The transformed values for all the batches along with their results are shown in Tables 3 and 4. The dependent variables (disintegration time, DT; friability, F; and drug release, Q5) obtained at various levels of the 3 independent variables (X1, X2, and X3) was subjected to multiple regression to yield a second-order polynomial equation. The coefficient values obtained are shown in Table 5. Among the 3 independent variables, regression analysis indicate that coefficients b13, b23, b11, and b33 for DT; b13, b23, b22 and b33 for F, and b12, b23, b11, and b22 for Q5 (p ≤ 0.05) were insignificant in predicting DT, F, and Q5. Hence, these terms were omitted from the full model to obtain a reduced second-order polynomial equation by multiple regression of DT, F, and Q5 and the significant terms (p ≤ 0.05) of reduced model equation. The value of the correlation coefficient (R2) of the polynomial regression equation was greater than 0.99, which is near to 1, thus indicating a good fit for all the dependent variables. The results of analysis of variance (ANOVA) are shown in Table 6. The DT, F, and Q5 values measured for the various batches showed wide variations. The results indicate that DT, F, and Q5 data were strongly affected by the variables selected for the study. The main effects of X1, X2, and X3 represent the average result of changing one variable at a time from its low to high level. The interaction terms (X1X2, X1X3, X2X3, X1X1, X2X2, and X3X3) show how the DT, F, and Q5 changes when two variables are simultaneously changed. The negative coefficients for all 3 independent variables (X1, X2, and X3) indicate a favourable effect on the disintegration time, while the positive coefficients for the interactions between 2 variables (X1X2, X1X3, X1X1, X2X2, and X3X3) indicate an unfavorable effect on the disintegration time. The positive coefficients (X1 and X3) for the independent variables show an unfavourable effect on friability, while the negative coefficients for the interactions between 2 variables (X2X3, X3X3, and X1X1) imply a favourable effect on the parameter (friability). The positive coefficients for independent variables (X1, X2 and X3) point to a favorable effect on Q5, but the negative coefficients for the interactions between two variables (X1X1 and X2X2) indicate an unfavourable effect on drug release (Q5). DISCUSSION Combinational effect on disintegration time The results of multiple linear regression analysis (full model) reveal that on increasing the amount of either camphor or crospovidone, a decrease in disintegration time was observed. When a higher amount of camphor was used, higher porosity was expected for the tablets. Due to the porous network of the tablet, water uptake increased and thus disintegration was facilitated. When a higher amount of superdisintegrant (crospovidone) was incorporated, wicking was enhanced, leading to a decrease in the disintegration time of the tablets. Addition of β-cyclodextrin might have enhanced the swelling of the tablet due to increase in the absorption of the medium. The combined effect of porous structure, wicking and swelling would have rendered the tablets very rapidly disintegrating. Combinational effect on friabilityAn increase in the concentration of camphor led to an increase in friability because the coefficient, b1, bears a positive sign. When a higher amount of camphor was used, more porous tablets, which showed mechanical weakness, were produced. Increase in the concentration of crospovidone resulted in decreased friability because b2 bears a negative sign. Thus, addition of crospovidone to the tablet formulation made it less friable. Tablets with low friability (≤ 0.6 %) may not break during handling, packaging and/or shipping [15]. Crospovidone is known to produce mechanically strong fast-dissolving tablets [15]. Combinational effect on drug releaseThe coefficient, b3, bears a positive sign which shows that increase in the concentration of β-cyclodextrin would also result in a rise in the dissolution of the drug. As indicated by the dissolution data for the physical mixture of the drug and βcyclodextrin, increased wettabilty and dispersabilty of the tablets led to improvement in drug dissolution. The effect of β-cyclodextrin on wettabilty can be attributed to the enhanced surface area available for dissolution due to reduction in interfacial tension between the drug and the dissolution medium. During the dissolution studies, it was noted that the tablet formulation containing β-cyclodextrin sank rapidly, unlike the tablets without β-cyclodextrin which remained floating on the surface of the dissolution medium for a period of time. In fact, both the impartation of a porous structure to the tablets by camphor and the wicking property induced by crospovidone are responsible for enhanced drug dissolution. The coefficients for drug release, b1 and b2, possess a positive sign which shows that they also facilitated the dissolution of the drug. Optimization of fast dissolving tablet formulationThe optimization of the fast-dissolving tablet formulation was targeted to achieve a disintegration time (DT) of 30 s, friability of 0.6 % and drug release of 90 % (at the end of 5 min). The optimized amounts, obtained with the aid of software, are shown in the surface response prediction curves in illustrated in Figure 1. A checkpoint batch (PMT) was prepared at X1 = -0.41 level; X2 = 0.61 level and X3 = 1.00 level at which DT was 30 s, friability 0.599 % and drug release (in 5 min) 88.13 %. The desirability of the optimization process was 0.989 which is near to unity. From the full model, it was found that the friability of the checkpoint batch, PMT, was 0.594 ± 0.007 %, disintegration time 31.00±0.73 s, drug release (at the end of 5 min) 89.5 ± 1.9 %, hardness 3.0 ± 0.2 kg/cm2, and wetting time 39 ± 0.09 s. As shown in Table 3, the optimized batch, PMT, indicates that the results were as expected. Thus, the statistical model is mathematically valid. Compared with the experimentally optimized preparation, the observed responses are in close agreement with the predicted values of the optimized formulation, thereby demonstrating the feasibility of the optimization procedure used in developing promethazine theoclate fast-dissolving tablet formulation. CONCLUSIONOptimization of fast-dissolving tablet formulation of promethazine theoclate using 33 factorial design was achieved in this study. The amount of independent formulation variables, camphor, crospovidone, and βcyclodextrin showed a significant effect on disintegration time and friability as well as the drug release characteristics of the fastdissolving tablets. The experimental design provided a better understanding of the effect of formulation variables on the quality of fastdissolving tablets containing the hydrophobic drug. The optimal batch exhibited a disintegration time of 31 s, friability of 0.06 % and drug release (within 5 min) of 89 %. Thus, by adopting a systematic formulation approach, an optimum point can be reached in the shortest time with minimal efforts. REFERENCES
Copyright 2010 - Tropical Journal of Pharmaceutical Research The following images related to this document are available:Photo images[pr10059t3.jpg] [pr10059t6.jpg] [pr10059t4.jpg] [pr10059t5.jpg] [pr10059t2a.jpg] [pr10059t1.jpg] [pr10059f1.jpg] [pr10059t2.jpg] [pr10059t2b.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}