 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Tropical Journal of Pharmaceutical Research, Vol. 9, No. 6, November-December, 2010, pp. 533-539 Research Article Formulation and In Vitro Evaluation of pH-Sensitive Oil-Entrapped Buoyant Beads of Clarithromycin GK Tripathi* and S Singh Industrial Pharmacy Laboratory, Department of Pharmacy, Saroj Institute of Technology & Management Lucknow, India-226001 *Corresponding author: E-mail: orgpharm@gmail.com; Tel: 09415745934 Received: 19 April 2010 Revised accepted: 14 October 2010 Code Number: pr10064 Abstract Purpose: To develop pH-sensitive controlled release formulation of clarithromycin
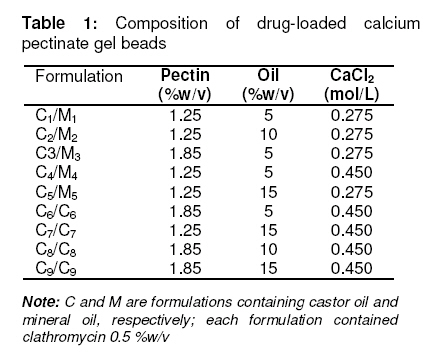
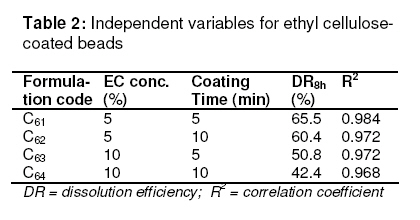
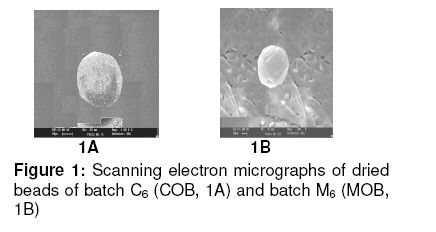
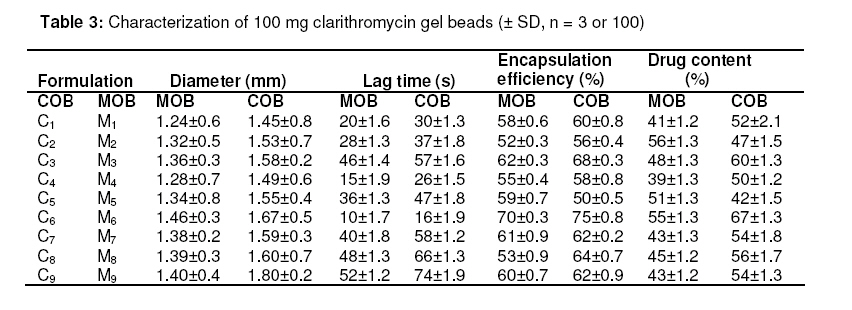
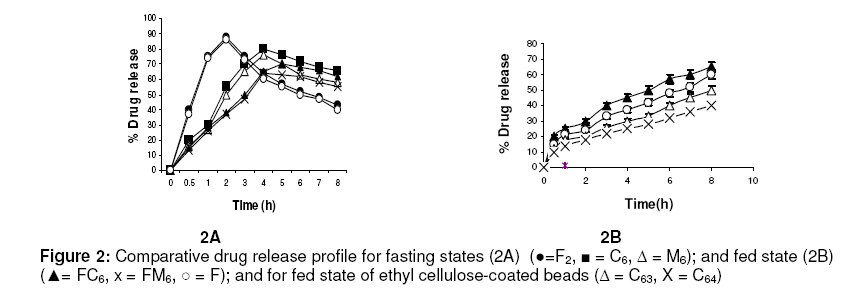
in oil-entrapped calcium pectinate microgel bead. Keywords: Clarithromycin; Calcium pectinate bead; Gastric residence time; pH-sensitive; Ethyl cellulose; Oil-entrapped INTRODUCTION The oral route has attracted special attention for the delivery of anti infective agents that are needed to produce local action in the gastrointestinal tract. This route of administration usually shows high compliance by patients due to ease of administration. A search of the scientific and patent literature reveals an increased interest in novel dosage forms for the targeting of different parts of the gastrointestinal tract for drug retention at the site of action for a predetermined time [1,2]. This approach is especially most attractive for the delivery of anti-infective agents for the targeting of local microbial lesions inside gastrointestinal tract and promises to provide a more effective cure of such infectious lesions than conventional dosage forms. This is because a major problem frequently encountered with conventional oral dosage forms is the inability to localise drug release in the stomach and proximal portion of the small intestine [3,4]. Floating drug and bioadhesive drug delivery systems are widely used techniques for gastroretention [5,6]. Clarithromycin is a semi-synthetic macrolide antibiotic derived from erythromycin. It is primarily bacteriostatic and exerts its antimicrobial effect by the inhibition of protein synthesis on bacterial ribosome [7,8]. Pectin is a colloidal polygalacturonic acid in which some of the carboxylic groups are esterified with methyl groups. The main constituent of pectin is D-galectouronic acid [9,10]. This low methoxy polysaccharide polymer, with a degree of esterification < 50 %, can form rigid gels in the presence of calcium ions or other multivalent cations which crosslink the galacturonic acid chains of pectin to yield hydrogels that are stable at low pH. Pectin can reduce interfacial tension between an oil phase and a water phase and is efficient for the preparation of emulsions [11]. The objective of this work was to develop a gastroretentive, multiple-unit, controlled release formulation of clarithromycin that would achieve continuous release of the drug in the gastric region and thus be useful for complete termination of microbial infection at gastric sites. EXPERIMENTAL Materials Clarithromycin was obtained as a gift from Ranbaxy Laboratories, Devash, India. Lowmethoxy pectin, with a degree of esterification of 35 %, and ethyl cellulose, were obtained from S.D. Fine Chemicals, India. Light mineral oil and castor oil were supplied by Central Drug House, India. Preparation of calcium pectinate beadsThe gel beads were formulated using a 23 factorial design. The effect of concentration of the oils (castor and mineral oils), pectin and calcium chloride were fixed in the formulation as independent variables. The effect of the dependent variables in the formulation was investigated in terms of bead diameter, floating lag time and encapsulation efficiency. The composition of eighteen batches of the drugloaded calcium pectinate beads is given in Table 1. Oil-entrapped calcium pectinate gel beads were prepared by ionic gelation method. The drug was dispersed in varying concentrations of aqueous solution of pectin (1.25 -1.85 %) with continuous stirring until a uniform dispersion containing 0.5 % of the drug was obtained. The mixture was emulsified with either mineral oil or castor oil using a Silverson emulsifier (Hicon, India) at a constant stirring rate of 500 rpm for 5 min. The resulting drug-loaded emulsion was dropped through a 21G syringe needle separately into 100 mL of 0.275 -0.45 mol ml -1 of calcium chloride solution and stirred with a magnetic stirrer to improve the mechanical strength of the beads and to prevent their aggregation. Formation of small microgel beads of clarithromycin based on either castor oil (COB) or mineral oil (MOB) occurred after 5 min of curing. The beads were washed with distilled water, collected by filtration throughWhatman filter paper no. 1 and dried in a tray dryer at 40 °C for 6 h. Coating of gel beadsThe selected gel bead formulations were coated with ethyl cellulose (EC) in a 22 factorial design (see Table 2) for optimization. The coating formulation was 5 –10 %w/v ethyl cellulose (EC) solution in acetone and coating time was 5 -10 min. The gel beads (2 g) were placed in a fluidized bed dryer (TG 100, Retsch, Germany) and the fluidized beads were sprayed with the coating solution for a period of 5 or 10 min at an air inlet speed of 220 m/s at room temperature. The beads were dried at room temperature for a period of 24 h when the solvent evaporated, leaving a film coat on the gel beads. Size and morphologyThe particle size of the beads were determined in three sets using an optical microscope (Model BH-2, Olympus, Japan) fitted with a stage micrometer. Twenty dried beads were measured for the calculation of mean diameter. The external and internal morphology of the beads were studied by scanning electron microscopy (SEM). In this assessment, the beads were first coated with gold palladium under argon atmosphere using a gold sputter module in a high vacuum evaporator. The coated samples were then observed with SEM. In vitro floating studyIn vitro floating test was performed using a USP 24 dissolution apparatus II in 500 mL of phthalate buffer solution (pH 3.4) with the medium temperature kept at 37 ± 0.5 °C. The floating beads (1 g) were placed in the dissolution medium agitated with a paddle at 50 rpm. After agitation, the beads that floated on the surface of the medium and those that settled down at the bottom of the flask were recovered separately. Lag time (the time taken for the beads to float at the surface of the medium) and floating behaviour were observed for up to 12 h [12]. Determination of drug-loading and encapsulation efficiencyAccurately weighed (100 mg) grounded powder of beads was soaked in 100 ml phosphate buffer (pH 7.5) and allowed to disintegrating completely for 4 h [13]. The resulting dispersion was sonicated using a probe sonicator (UP 400 s, Dr. Hielscher GmbH, Germany) for 30 min and then filtered through a 0.45 µm filter. The polymeric debris was washed twice with fresh phosphate buffer to extract any adhered drug and drug content was determined spectrophotometrically at λmax of 353 nm against a constructed calibration curve. The encapsulation efficiency (EE) was calculated according to the relationship in Eq 1. EE (%) = (C/T) x 100 ……………….………. (1) where C is the calculated drug content and T is the theoretical drug content. In vitro drug releaseIn vitro dissolution studies were performed for the gel beads using USP 24 dissolution test apparatus II (basket type) [13]. Accurately weighed 50 mg amount of the bead (containing 19 – 21 mg of active drug) dropped in 900 ml of simulated gastric fluid (SGF, fasting state, pH 1.2; prepared by dissolving 2 g of sodium chloride, 3.2 g pepsin, and 6.8 ml of hydrochloric acid in double distilled water to 1 L), or fed state (phthalate buffer solution, pH 3.4) maintained at 37 ± 0.5 ºC and stirred at a speed of 50 rpm. At different time intervals over a period of 8 h, a 10 mL aliquot of the medium was withdrawn and replenished with an equivalent volume of plain dissolution medium. The samples were filtered, suitably diluted and analyzed at a wavelength of 353 nm using a UV-visible spectrophotometer (Shimadzu). The drug release data were corrected for drug loss during sampling and degradation at acidic pH. All the tests were carried out in triplicate. Additionally, an experimental batch containing 10 mg clarithromycin and lactose (q.s.) filled into an empty capsule shell (#2) was used as a reference formulation. Kinetic release evaluationTo investigate the mode of drug release from the microgel beads, the release data were analyzed with various release kinetic models (zero order, Higuchi and Korshmaer-Peppas) were applied to elucidate their mechanism of drug release in the fed state [14-16]. The analysis of the dissolution data was carried out using Eqs 2 – 4 for zero order, Higuchi and Korsemeyer-Peppas models, respecttively. Mt = M0 + K0t …………………...……… (2) where Mt is the amount of drug dissolved in time t, M0 is the initial amount of drug, K0 is the zero order release constant and KH is the Higuchi rate constant. Mt/M∞ is the fraction of drug release at time t, k is the release rate constant, and n is the release exponent indicative of the mechanism of release. Statistical analysisThe results were expressed as mean ± SD (standard deviation). Statistical evaluation of the data was performed using analysis of variance (ANOVA) and, depending on the outcome of ANOVA, Dunnett’s multiple comparison test was also applied. Statistically significant difference between the means of batches was set at p < 0 .05. RESULTSThe scanning electron micrographs (SEM) of the dried microgel beads, C6 and M6, are shown in Figure 1. Gel beads prepared from mineral oil (MOB) were white, translucent and rigid, while castor oil-based gel beads (COB) were off–white, translucent and elastic. The diameter of MOB varied between 1.24 ± 0.60 and 1.46 ± 0.30 mm while that of COB was between 1.45 ± 0.80 and 1.80 ± 0.20 mm. Floating lag time (the time taken for the beads to float at the surface of the medium) was 10 –52 s and 16 -74 sfor MOB and COB, respectively (Table 3). Encapsulation efficiency was highest for batch C6 (75.0 ± 0.8 %) and batch M6 (70.0 ± 0.3 %) while drug content was 55.0 ± 1.3 % and 67.0 ± 1.3%, respectively, for the batches. In vitro drug releaseThe dissolution data are shown in Fig 2. The results indicate that 90.1 ± 2.5 % of the pure drug (batch F) dissolved in 2 h in fasting state (pH 1.2) while for the fed state (F2, pH 3.4), the figure was 87.5 ± 2.5 %. In 4h, drug release from batch C6 was 80.0 ± 2.4 % (fasting state) and from batch M6 76.0 ± 2.7 % (fed state) in simulated gastric fluid (SGF). For the EC-coated beads (batch C61), a maximum dissolution efficiency of 65.5 % was attained in 8 h, as shown in Table 2. Drug release from the optimized bead formulations C6 and M6 followed the Higuchi (R2 = 0.9841, n = 0.41) and Peppas models (R2 = 0.9827, n = 0.39), respectively. Correlation coefficient (R2) of the coated batches (see Table 2), based on zero order release kinetics, ranged from 0.968 – 0.984. DISCUSSION Spherical gel beads were formed instantaneously when emulsion was dropped into calcium chloride solutions. Gelation occurred due to intermolecular cross-linking between the divalent calcium ions and the negatively charged carboxyl groups of pectin. Pectin promoted the emulsification of the mixture of water and oil phases during homogenization and the resulting oil droplets were dispersed in calcium crosslinked network of the formulation. The diameter of the beads increased significantly (p < 0.05) as polymer concentration increased; this could be attributed to the increase in the microviscosity of the polymeric dispersion, eventually leading to the formation of larger beads. Larger size beads were also formed as the concentration of calcium chloride increased. This may be due to excess calcium ions causing possibly all the crosslinking sites in the polymer to be fully utilized and resulting in larger but weaker and flexible gel beads. Buoyancy is an important factor in sustained drug delivery to the gastric region. All the beads floated on simulated gastric fluid for up to 12 h. Increase in the calcium chloride content of the beads resulted in a decrease in floating lag time. due to increase in the porosity of the gel beads. Floating lag time also rose as the concentration of oil in the formulation increased and this can be attributed to flocculation of the oil globules which might also have coalesced to produce large droplets. The encapsulation efficiency of the beads rose as polymer concentration increased due to the availability of excess polymer which ensured that the drug was optimally entrapped. On the other hand, encapsulation efficiency decreased with increase in calcium chloride concentration because excess Ca2+ would have the effect of weakening the polymer gel structure and strength, thus leaving it more porous and limiting its capacity to trap the drug. Drug release from the beads (as shown in Fig 2A) was characterized by an initial phase of rapid release (‘burst effect’) due to the presence of clarithromycin on the bead surface since the drug exhibits good solubility at low pH [17].. However, release thereafter slowed down due to the obstruction of drug diffusion by pectin -Ca2+ ions crosslinks. The release exponent (n) value of 0.045 suggests a diffusion-based release mechanism [18]. However, the dissolution profiles of the coated beads (batch C62, C62 ,C63 and C64) were best fitted to the zero-order kinetic model (Fig 2B ) . Formulation batch C61 was considered the optimized gastroretentive controlled-release floating gel bead for clarithromycin, as it showed the lowest release. CONCLUSIONThe developed oil-entrapped gel beads showed good floating and controlled drug release properties at simulated acid pH conditions of the stomach. Therefore, it may be capable of delivering clarithromycin to stomach sites, thus opening up the possibility of targeting the drug to gastric sites for the treatment of microbial infections such as that caused by H. pylori. ACKNOWLEDGEMENTThe authors are grateful to IIT Roorkee, India for making available SEM facilities for the work. Special thanks also to S.K. Agrawal, Administrative Director of Saroj Institute of Technology and Management, Lucknow, India, for providing various facilities for the study. REFERENCES
Copyright 2010 - Tropical Journal of Pharmaceutical Research The following images related to this document are available:Photo images[pr10064t3.jpg] [pr10064t2.jpg] [pr10064t1.jpg] [pr10064f2.jpg] [pr10064f1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}