 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
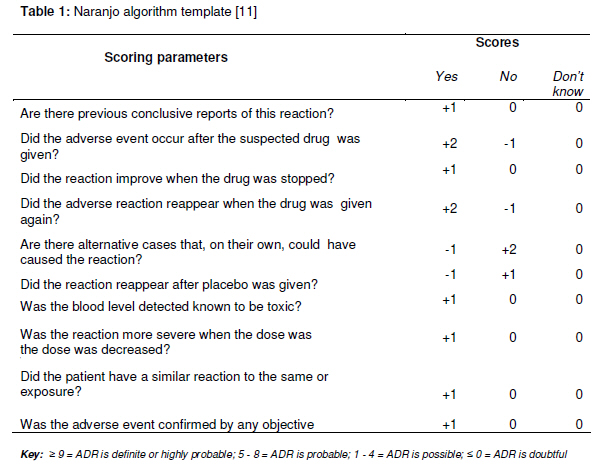
Tropical Journal of Pharmaceutical Research February 2011; 10 (1): 3-9 Opinion Article Approach to Publishing Adverse Event Case Reports in Biomedical Journals Kazeem A Oshikoya1,2Pharmacology Department, Lagos State University College of Medicine, PMB 21266, Ikeja, Lagos, Nigeria, and Academic Division of Child Health, The Medical School in Derby, Royal Derby Children’s Hospital, Uttoxeter Road, Derby DE 22 3DT, United Kingdom. *Corresponding author: Email: med_modhospital@yahoo.com, Tel: +44-796-128-4468 Recieved: 13 August 2010 Revised accepted: 28 November 2010 Code Number: pr11002 Abstract Case reports describing suspected adverse events of drugs and medical products are important for post-marketing safety monitoring. Such reports could help identify potential product-associated risks and serve as signals of possible events that may require further studies. They also serve as legal documents and have been used as evidence in “toxic tort” litigation. Lack of relevant details in the reports would render them of limited value and misleading. Deficiencies in the previously published adverse event case reports in some biomedical journals from developing countries clearly illustrate a need for guidelines. A properly documented report should provide details to enable readers make differential diagnoses, ascertain the causality of the reaction to the suspected drug, and provide pharmacological and biological explanations for the reaction. Authors should also report the suspected events to the National Pharmacovigilance Centre to ensure their inclusion in adverse drug reaction databases. Reviewers and journal editors should be well familiar with adverse drug event reporting guidelines to enable them weed out junk manuscripts. It would seem reasonable to include such guidelines in the instructions for authors, should a journal continue to publish case reports of adverse drug events. Keywords: Adverse drug events, Reporting and publishing, Biomedical journals, Developing countries Introduction Drug development is a long process that begins with animal studies, followed by human studies, and through clinical trials. Marketing drugs for general use begins when they are found to be unharmful to humans at therapeutic doses and their adverse effects are minimal. However, some rare adverse effects may not be detectable during the clinical trials; therefore post-marketing surveillance is instituted as a follow up to their recommended use. Drugs and medical products that include traditional, herbal and complementary medicines, vaccines and other biologicals and devices may sometimes be harmful to the body when used according to recommendation, thus it becomes necessary to monitor their safety after they have been marketed. Publication of case reports describing suspected adverse events of drugs and medical products is important for post-marketing safety monitoring [1]. Such reports help identify potential productassociated risks and serve as signals of possible events that may require further studies [1]. A properly documented adverse event report should create awareness to clinicians and avert possible consequences that may be serious if the adverse effect was realised late. Hypotheses can also be generated from such reports, between the suspected products and the associated events that would enable policy-makers to have a complete understanding of the benefit-risk potentials of the drug or medical product. Adverse case reports are also legal documents and have been used as evidence in “toxic tort” litigation which is increasingly common in the developed countries [2]. Adverse event case reports are usually scientific publications originating from healthcare professionals who, during their duties of caring for patients, suspect potentially unusual or untoward events that they consider to have a causal relationship to a drug or medical product. However, the average medical doctor is not a trained researcher and may not likely know how to properly report an adverse drug event [2]. The quest for academic promotion has been the drive for most doctors to publish case reports and this has contributed significantly to under-reporting of adverse drug reactions (ADRs) to the drug regulatory authorities [3]. By obligation, it is necessary to report the case or case series to the National Pharmacovigilance Centre. Pharmacovigilance experts have expressed concerns over the considerable variation in the completeness of published adverse event reports in international peer-reviewed journals [4-7]. Case reports writing has gone beyond mere compilation of a brief account of the patients’ history, describing the odd features, reviewing similar cases that were previously reported, and offering speculative theory about the causal relationship between the suspected drug and the event [2]. Such reports would be of limited value and often misleading as they lack the relevant details. An analysis of a recently published case series report of toxic epidermal necrolysis to oral dihydroartemesinin in Nigeria revealed that information about the relevant drug exposures, the clinical decision making processes, the possible alternative aetiologies and the causality assessments were all lacking [8]. Given the deficiencies of the adverse event case reports published in some biomedical journals from developing countries, a review that is aimed at improving adverse event case reporting by authors, serves as a guide to editors of biomedical journals and advocate for the development of standard local guidelines published in some biomedical journals from developing countries, a review that focuses on the guidelines for publishing high quality case reports was considered necessary. Guidelines for Authors Authors should aim at properly describing a case of adverse drug event by providing sufficient details for differential diagnoses, provisional assessment of a causal association between a suspected drug and the event, and a reasonable pharmacological or biological explanation for the cause-effect [1]. The International Society for Pharmacoepidemiology (ISPE) and the International Society of Pharmacovigilance (ISoP) has jointly recommended the following data elements when writing an adverse event report for publication [1]: TitleIt is required that the title should be consistent with the content of the report. This was rather lacking in the adverse event report to dihydroartemesinin published in a Nigerian medical journal [8]. While the title suggested an adverse event, the discussion focused only on managing the fatal outcome. Patient’s information Demographics: It is necessary to state the age and gender of the patient and highly desirable to indicate the weight, especially if it was a child. Other relevant demographics include height, race and ethnicity, obstetrical status, body mass index, social life, and occupation. Current health status: It is highly required to describe the disease or symptoms being treated initially with the suspect drug, and state its duration and severity. Medical history: Co-morbidity or pre-existing diseases may be the underlying risk factors. Therefore, it is very necessary to document all medical history relevant to the adverse event and prior drug or medical product exposures. Previous therapy of active disease, alcohol, tobacco, and substance or illicit drug abuse history should also be sought and documented. Relevant social circumstances, family history, and drugs taken by household members should be sought if relevant to the reported adverse event. This is because if genetic factors were suspected as cause of the adverse event, the suspected drug may be a cause if the relatives of the patient had once been affected. Physical examination: The physical or laboratory findings should be detailed. Both abnormal and normal findings should be reported. Baseline laboratory results with normal range of values should also be provided. If off-label or unlicensed drug was suspected, reasons for its use should be documented. Event outcome: Some adverse drug events may result into short or prolonged hospitalisation, life threatening conditions, significant morbidity or mortality, or poor quality of life after recovery. These outcomes, as well as presence or absence of death, need to be documented. Drug history Identification and characterisation: Suspected drug(s) must be identified by generic name. Brand names may, however, be mentioned with strength or dosage units, and the manufacturer. Herbal products should be described by their herbal ingredients, plant parts, and types of preparation such as crude herbs or extracts, type and concentration of extraction solvent used and drug-extract ratio.Dosage: Exact or approximate dose of the suspect drug and others used in the case of multiple drug therapies, as well as the serum or other fluid drug concentrations should be mentioned. Duration of therapy should indicate the start and stop dates. If the suspect drug was later recommenced, the restart date should be mentioned. Route of drug administration: This may be oral, buccal, sublingual, intranasal, intramuscular, intravenous, rectal or topical. The route, as well as adherence to recommendation, should be mentioned. Drug-reaction interface: This implies the therapy duration before the adverse event. This should include the first dose-event, last dose-event, and last dose-resolution intervals.Concomitant therapies: History of drug therapy should focus beyond the immediate therapy preceding the adverse event [9]. The history of drug exposures should span, at least, a month before onset of the adverse event [9,10]. In the case of multiple drug therapies, the potential contribution of the concomitant therapies should be assessed. These therapies would include prescription and non-prescription medicines such as herbal or complementary medicines. Table 1: Naranjo algorithm template [11] Adverse EventThe adverse event and its severity must be described to details. Onset date and duration of the event should be mentioned. Case definition is necessary if more than one patient is involved. Adverse event should be defined according to conference or established/validated criteria. Several methods have been used to assess the causality of an adverse drug event [11,12]. All the methods do incorporate part of the Bradford-Hill’s criteria [13] and none has been shown to be more superior to any of the others. Although the validity and reproducibility of the Naranjo algorithm (Table 1) has been criticized [14], it is the most widely used [11]. Reporting adverse events to medications with an established causality would require the author(s) to submit an objective causality assessment along with the manuscript. The outcome of the event should be mentioned as earlier highlighted. Specific treatments for adverse event that were instituted should be mentioned. DiscussionThis should include presence or absence of evidence that supports a causal relationship of the suspected drug to the adverse event including timing, de-challenge and rechallenge. If it was impossible to carry out these assessment steps, authors should give explanations [11]. Diagnostic procedures performed to confirm the final diagnosis should be clearly stated. Plausible pharmacological or biological explanation(s) for the adverse events should be provided. Discussion of case series reports should follow the recommendations of Edwards et al [15]. Previously published reports of the adverse event in biomedical journals or product labelling should be discussed. Prior reports to regulatory agencies should also be discussed. Competing explanations should be assessed in the discussion. Any progress or planned clinical trials of the adverse event should be discussed. Team management and authorship contributionOptimal management of adverse drug events requires a team approach comprising of physician(s), clinical pharmacist, clinical pharmacologist, expert in pharmacoepidemiology and pharmacovigilance. Poor knowledge and perception of ADRs reporting has been reported among physicians in Nigeria [16-18]. Also, undergraduate and postgraduate teaching of pharmacovigilance and risk perceptions has been reported to be inadequate [19]. The ill-equipped physicians may likely be confused on what to report. It is, therefore, advisable to involve other experts in the patient management and manuscript preparation. A clinical pharmacist or pharmacologist could help on drug information, selection of alternative drugs, and causality assessment. Guidelines for Reviewers Given the peculiarity of adverse event case reports, reviewers should be, at least, two in numbers. One should be an expert in the primary disease for which the patient was treated and the other an expert in pharmacoepidemiology or pharmacovigilance. Dearth of these experts in many developing countries would necessitate making an online search for local experts and if possible seek expert opinion from developed countries. Reviewers should be guided by the guidelines earlier set out for authors and ensure that author(s) adhere strictly to the journal’s instructions. In those adverse event case reports where author(s) had performed and reported a causality assessment, reviewers should ascertain an association between the suspect medicine and the adverse event. They should be provided with the Naranjo algorithm [11] or other validated criteria for causality assessment [12]. This will enable reviewers to give a numerical score to individual report or consistently rate the report as “unlikely”, “possible”, or “highly probable”. The grading process would add no further data, and would neither validate nor refute author’s report. Rather, it would make the merits and faults of the case report stand out clearly for the editor to see. Despite the ISPE/ISoP guidelines, individual adverse event case reports may have their limitations. Reviewers should ensure that such limitations are clearly mentioned and critically discussed by the author(s). Reviewers may recommend to the editor other experts they consider competent in pharmacoepidemiology or pharmacovigilance to undertake peer-review of adverse event case reports. Guidelines for Editors Most biomedical journals from developing countries have no requirements for publishing adverse event reports. Therefore, guidelines are required when reviewing such case reports at the editorial level. This would enable the editors to halt dissemination of junk scientific information. Individual genetic variation and differences in disease and drug use pattern of developing and developed countries may result in variations of adverse events to specific medicines. Thus, local guidelines for publishing adverse drug events may be required by authors and reviewers. Such local guidelines should incorporate some of the ISPE/ISoP guidelines and be applicable to the editors of biomedical journal. The proposed guidelines for authors and reviewers are applicable to the editors. It would seem reasonable to include a validated and appropriate scale for causality assessment of the likelihood that the events were drug-related in the instruction for authors. A summary of the local guideline should be included in the instructions for authors and be strictly enforced as practiced by the Journal of American Family Physician [20] and the Annals of Pharmacotherapy [21]. Evidence that the adverse event has been reported to the National Pharmacovigilance Centre, for countries with such a reporting system, should form an important criterion for an editorial review before such manuscript is assigned a reviewer and, if possible, authors should provide the report number. This important step is strongly recommended by the ISPE and ISoP [1]. Roles of Policymakers The National Pharmacovigilance Centre, national and drug regulatory agencies, various pharmacology and pharmacy regulatory bodies, experts in clinical pharmacology, pharmacoepidemiology and pharmacovigilance, herbal medicine practitioners, and editors of biomedical journals should work together to develop local standard guidelines that would incorporate the ISPE/ISoP suggestions. Editors of biomedical journals are urged to apply such guidelines to case reports submitted for publication. The regulatory body in charge of medical education in each developing country should consider circulating the local standard guidelines to all medical, pharmacy and nursing schools for incorporation into the relevant curricula that address the detection, evaluation, and reporting of suspected drug or other medical product adverse events. ConclusionAdverse event case reports published in biomedical journals from developing countries may be an appropriate way of enhancing pharmacovigilance. Detailed reporting is highly required in order to avert misinformation and dissemination of wrong signals to clinicians, pharmaceutical industries and drug regulatory agency. Conflict of Interest The author is a member of the Adverse Drug Reaction Monitoring Committee of the Lagos State University Teaching Hospital (LASUTH), Ikeja, Lagos, Nigeria and a member of the International Society for Pharmacoepidemiology. > > > > References
The following images related to this document are available:Photo images[pr11002t1.jpg] |
| |||||||||

{kind=link}