 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
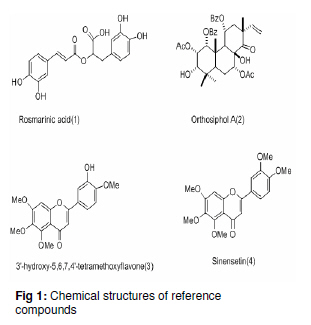
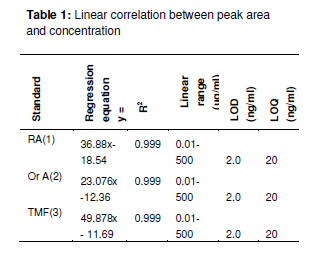
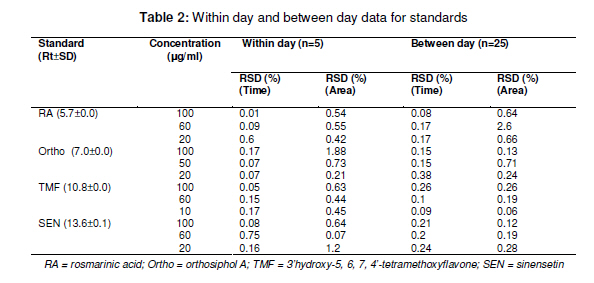
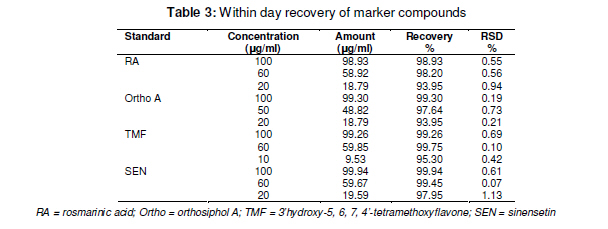
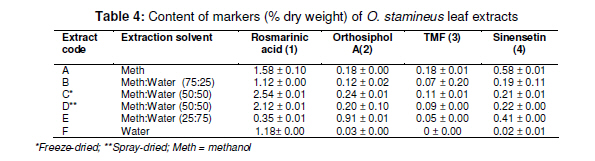
Tropical Journal of Pharmaceutical Research February 2011; 10 (1): 97-103 Research Article Simultaneous Analysis of Bioactive Markers from Orthosiphon Stamineus Benth Leaves Extracts by Reverse Phase High Performance Liquid Chromatography Mohammad Jamshed Ahmad Siddiqui* and Zhari Ismail Pharmaceutical Chemistry, School of Pharmaceutical Sciences, UniveFrsiti Sains Malaysia, 11800 Minden, Penang, Malaysia *Corresponding author: E-mail: siddiquijamshed@hotmail.com, siddiquimj@gmail.com; Tel/Fax: 0060-46563443 Received: 10 April 2010 Revised accepted: 14 November 2010 Code Number: pr11015 Abstract Purpose: To develop a reverse phase high performance liquid chromatography (RP-HPLC) method for the analysis Keywords: Orthosiphon stamineus, Orthosiphol A, Rosmarinic acid, Sinensetin, Isocratic, Quantification, HPLC Introduction Orthosiphon stamineus (OS), Benth, (Lamiaceae) is one of the most useful traditional medicinal herbs cultivated in South East Asia, particularly Malaysia and Indonesia. Being popularly known as Java tea, this herb is widely used in traditional medicine to treat many ailments due to its activities such as diuretic, urolithiatic, antiinflammatory, anticholestatic, analeptic, antirheumatic as well as antidiabetic [1,2]. The leaves of O. stamineus have been used as a diuretic in the form of infusions in a wide variety of kidney and bladder pathologies, especially kidney stones, pain in the bladder with frequent urination, as well as accumulation of uric acid crystals in joints owing to elevated blood uric acid levels [3-5]. Technological strides in chromatographic and spectroscopic methods have brought tremendous influence on the isolation and structural elucidation of a variety of medicinal plants constituents. The recent surge of interest in O. stamineus has led to the isolation and identification of several classes of bioactive compounds such as flavanoids, diterpenes, triterpenes, saponins, sterols organic acids, caffeic acids derivatives and chromenes [69]. Hussain and associates isolated betulinic acid, 16-β-hudroxybetulinic acid and rosmarinic acid from this plant in Malaysia [10,11]. Numerous studies have also been performed to investigate the biological effects of O. stamineus as antioxidant, diuretic, antifungal as well as in alleviating hyperglycaemia and improving lipid profile in diabetic rats [3,9,12]. Earlier studies suggest that the flavones, sinensetin and 3’-hydroxy-5, 6, 4’-tetramethoxyflavone isolated from O. stamineus exhibited diuretic activity in rats which may be partially due to its lipophilic flavone content [12]. Recently, antiangiogenic activity has been attributed to the plant [13,17]. The aim of this study is to develop a simple and sensitive method for the simultaneous quantification of bioactive markers which can be used for the quality control of herbal products derived from O. stamineus plant. Experimental Plant material Orthosiphon stamineus plant was cultivated and propagated under controlled conditions in a joint venture with USM-UNIMAP at Titi Tinggi, Perlis, Malaysia. A voucher specimen (no. 11009) was deposited at the herbarium of the School of Biology, Universiti Sains Malaysia. The leaves were collected in the month of July and August 2009 and pulverized into a fine powder using a milling machine (Retsch GmbH, Germany) Chemical and reagents HPLC grade methanol, acetonitrile, tetrahydrofuran and ortho-phosphoric acid were purchased from Merck, Germany. Reference standards -sinensetin, 3’-hydroxy5, 6, 7, 4’-tetramethoxyflavone (TMF) and rosmarinic acid -were purchased from Indo Fine Chemical Company, Hillsborough, USA. Deionised water for HPLC was prepared using ultra pure water purifier system (Elgastat, Bucks, UK). Chemicals and solvents (analytical grade) for isolation of orthosiphol A were purchased from Merck Germany, while Silica gel 60 F254 preparative TLC and analytical TLC were purchased from E Merck, Germany. Preparation of extract The powdered crude drug extracted with water or methanol or with the following methanol: water mixtures: 75:25, 50:50, 25:75, using a Soxhlet extractor (for 12 h) in the case of methanol and methanol:water extraction. For water extraction, hot maceration at 50 oC for 6 h was employed and then repeated thrice. In each case, the extract was bulked and concentrated in a rotary evaporator under vacuum, and then freeze-dried. The lyophilized extracts were kept in a freezer until used. Isolation of orthosiphol A The dried extract was re-dissolved in water:acetone (90:10) mixture and extracted with hexane, separating the upper layer of hexane with the aid of a separating funnel. The mother liquor was again extracted with dichloromethane and the lower layer of dichloromethane-rich extract was separated. Mother liquor again was extracted with nbutanol and the butanol-rich upper layer containing the extract was separated. Each of the extract type was concentrated and dried in a rotavapor under high vacuum. The dichloromethane-rich extract was adsorbed on silica gel and column chromatography was performed using a glass column (30 x 2.5 cm) packed with silica gel (70 -230 mesh), eluting it with hexane by increasing its polarity with ethyl acetate. Several fractions (fractions 2 and 4) were collected and monitored on TLC. Fraction 2 was again chromatographed with 2.5 % methanol in chloroform over silica gel (70 230 mesh). Further purification was carried out with preparative thin layer chromatography to yield a pure compound. Purity (95 %) of compounds was confirmed by HPLC (Agilent Technologies). The identification and characterization of isolated compounds were performed by comparing ultraviolet (UV), Fourier transform infrared (FTIR), mass spectrometry (MS) and nuclear magnetic resonance (NMR) spectra as described previously [8]. Sample preparation for HPLC analysis Samples of extract (100 mg each) were dissolved in 25 ml mixture of methanol: water (1:1), and sonicated for 10 -15 min. The contents were transferred to a 25 ml volumetric flask, made up to the 25 ml mark and filtered through a 0.45 µm filter (Whatman). In the same way, the reference compounds (about 5 mg each) were dissolved in 5 ml of methanol and then filtered through a 0.45 µm filter. The stock solutions were used for further dilutions. Samples were kept in freezer at -20 oC prior to analysis. Instrumentation and chromatographic conditions High performance liquid chromatography (HPLC, Agilent Technologies Series 1100 system) used was equipped with a degasser, an auto sampler, a column heater, quaternary pump and UV detector. The column (Nucleosil C18, 250 mm x 4.6 mm, 5 µm) was maintained at 25 oC and The injected sample (20 µl) was eluted with an isocratic mobile phase comprising of methanol: tetrahydrofuran:water (0.1% H3PO4) mixture in the volume ratio 55:5:40. Flow rate was 0.7 ml/min, separation time 30 min, and detection at 330 nm. Data acquisition was performed with the aid of ChemStation A.08.03. Standard calibration curves were established by plotting the peak area against concentration. The reference compounds used were: rosmarinic acid, orthosiphol A, 3’hydroxy-5, 6, 7, 4’-tetramethoxyflavone and 5, 6, 7, 4’, 5’pentamethoxyflavone or sinensetin (see Fig 1). Determination of limits of detection (LOD), limits of quantification (LOQ), and linearity The linearity of the calibration curves was evaluated by linear regression analysis and correlation coefficient (R2). Limit of detection (LOD) was established at a signal-to-noise ratio (S/N) of 3 while limits of quantification (LOQ) were established at a signal-to-noise ratio (S/N) of 10. LOD and LOQ were experimentally verified by six injections of each standard. LOD and LOQ) were evaluated by measuring the magnitude of analytical background after injecting the blank sample. Method validation The precision and accuracy of the method was performed by within day and between day run validations. Each standard curve was separately constructed on each day of analysis. The within day precision and accuracy were determined for each standard on three concentration with five replicates on a single day. The resulting retention time and peak area were used to calculate standard deviation and relative standard deviation (% RSD). The accuracy of the method was verified by recovery studies by spiking the standards at three different concentration levels. Accuracy was calculated from plot of the value of detection versus added amounts. Results Characterization and structure elucidation of isolated compound (2) Isolated compound (2) was identified as orthosiphol A and was used as reference compound for determination of crude extracts obtained from O. stamineus leaves. Orthosiphol A white amorphous solid (10 mg), Rf 0.3 [Silica gel, solvent Toluene: Acetone (85:15)]. LC-MS gave m/z 677 [M+H]; UV (MeOH) λ max: 230, 280, nm; FTIR (KBr): 3445, 2951, 1741, 1281, 1245, 780 cm-1 . 1H NMR (CDCl3) δ: 1.01-1.15 (9H, s, H17,H18& H19), 1.43 (3H, s, H20), 1.90 (3H, s, 2-Ac), 2.21 (3H, s, 7-Ac), 5.31 (1H,brd, J=2.6Hz, H1), 5.42 (1H,brt, J=3.2Hz, H2), 3.55 (1H, m, H3), 2.45 (1H, dd, J=10 and 4.5Hz, H5), 2.1-2.22 (1H, m, H6), 5.45 (1H, brt, J=3.2Hz, H7), 3.12 (1H, brd, J=5.7Hz, H9), 5.82 (1H, m, H11), 1.99 (1H, dd, H12), 2.54 (1H, dd, H12), 5.63 (1H, dd, H15), 4.82 (1H, d, H16), 4.86 (1H, d, H16), 7.21-7.78 (10H, t, 1&11-Bz), 2.20 (1H, d, 3-OH), 2.85 (1H, brs, 8-OH). 13C NMR (CDCl3) δ: 75.1 (C1), 65.8 (C2), 77.3 (C3), 38.9 (C4), 34.6 (C5), 21.2 (C6), 71.2 (C7), 76.9 (C8), 43.2 (C9), 44.1 (C10), 69.3 (C11), 39.0 (C12), 46.3 (C13), 209.1 (C14), 144.2 (C15), 112 (C16), 26.9 (C17), 22.9 (C18), 29.9 (C19), 15.9 (C20), 21.1 (2-Ac), 173 (2-Ac), 21.1 (7-Ac), 167.1 (7-Ac), 165 (1-Bz), 167 (11-Bz), 136 (1-Bz), 135 (11-Bz), 127.2 (1-Bz), 128.3 (11-Bz); LC-MS gave m/z 677 [M+H]. Spectral data were consistent with that reported for orthosiphol A [8]. Limits of detection (LOD) and quantitation (LOQ) Table 1 indicates the calibration, LOD and LOQ data for the standarda. The curve was linear over the entire concentration range investigated with correlation coefficient ranging from 0.9996 to 0.9999 and standard deviation less then ±5 %. LOD and LOQ for the standards were 2 and 20 ng/ml, respectively, at a signal to noise ratio of 1:10. Retention time, recovery and precision As Table 3 shows, the mean retention time for the markers -rosmarinic acid, orthosiphol A, TMF and sinensetin -was 5.6 ± 0.02, 6.5 ± 0.01, 9.9 ± 0.04, and 12.2 ± 0.09, respectively, while RSD was in the range of 0.06 to 2.4 which is less than 5%. Marker contents of O. stamineus extracts As shown in Table 4 indicates, all the analysed samples showed a wide range in marker contents. Methanol extract contained the highest amounts of sinensetin but also contained other markers. Methanol:water (1:1) extract was rich in rosmarinic acid while methanol:water (25:75) extract was rich in orthosiphol A. These results are in agreement with previous findings [12]. Discussion Some methods have previously been used for the analysis of O. stamineus leaf extract [12-15]. The focus of the present work is on anticancer markers for the purpose of preparing standardized extracts for the analysis of anticancer and antiangiogenic activity profiling [13,17]. Sahib and associates highlighted that role of antioxidants in the antiangiogenic activity of the methanol extract of O. stamineus [13]. The high contents of rosmarinic acid and sinensetin [12,14] in the methanol and methanol:water (1:1) extracts may be the reason for their antiangiogenic activity is higher for the methanol extract [13] than for the freeze-dried and spray-dried methanol: water (1:1) extracts [17]. The overlaid chromatographs (see Figure 2b) suggest that the developed method is best suited for flavonoids markers because the other prominent peaks at Rt of 18 -23 min may be other lipophilic flavonoids or may contain methyl ripariochromene compounds [15]. Conclusion The proposed method is simple, sensitive and specific for simultaneous determination of marker compounds, either qualitatively or quantitatively. This method can be used as a fingerprint profile for the standardization of both extractives and herbal medicines derived from the O. stamineus. Acknowledgement The authors would like to thank Universiti Sains Malaysia for providing fellowship and also the Ministry of Science and Technology, Malaysia for extending a grant for the work. References
© Pharmacotherapy Group, Faculty of Pharmacy, University of Benin, Benin City, 300001 Nigeria. The following images related to this document are available:Photo images[pr11015t4.jpg] [pr11015t2.jpg] [pr11015f2.jpg] [pr11015t1.jpg] [pr11015f1.jpg] [pr11015t3.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}