 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
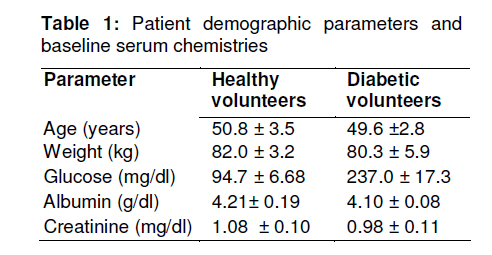
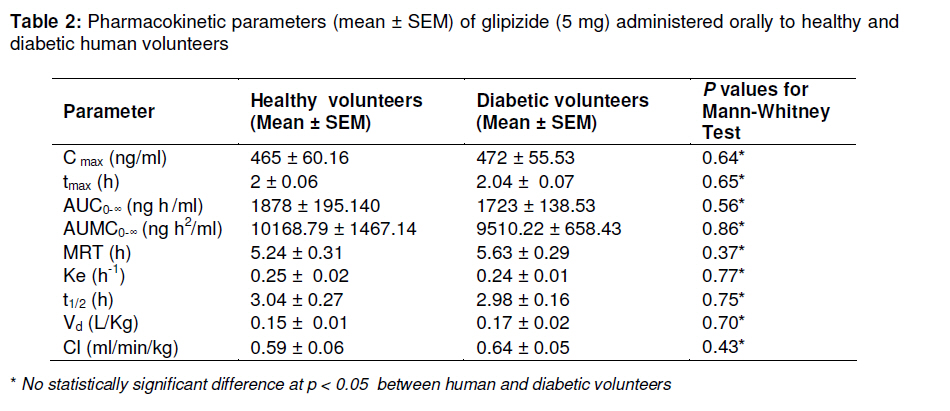
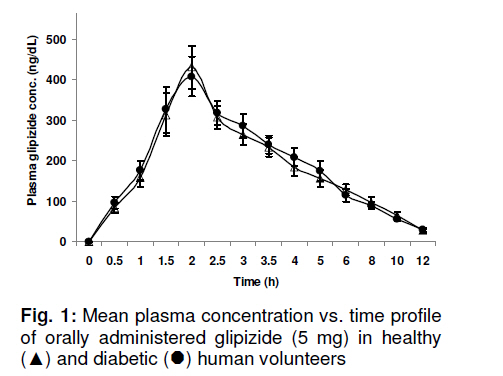
Tropical Journal of Pharmaceutical Research, Vol. 10, No. 2, April, 2011, pp. 147-152 Research Article Glipizide Pharmacokinetics in Healthy and Diabetic Volunteers Muhammad Atif1*, Mahmood Ahmad2, M Qamar-uz-zaman2, Muhammad Asif2, Syed Azhar Syed Sulaiman1, Asrul Akmal Shafie3, I Masood1, Usman Minhas2 and Najam Us-saqib4 1Discipline of Clinical Pharmacy, School of Pharmaceutical Sciences, University Sains Malaysia, 11800, Penang, Malaysia, Received: 13 September 2010 Revised accepted: 15 February 2011 Code Number: pr11021 Abstract Purpose: Disease state may contribute to alteration in drug pharmacokinetics. The purpose of this study was
to determine the effect of non-insulin dependent diabetes mellitus (NIDDM) on the pharmacokinetics of
glipizide.
Keywords: Glipizide, Bioavailability, Pharmacokinetics, Plasma, Reversed Phase-High performance liquid chromatography. INTRODUCTION Diabetes mellitus is not a single disease but a group of clinically heterogeneous disorders with glucose intolerance in common. It encompasses many causally unrelated diseases and includes many different etiologies of disturbed glucose tolerance [1,2]. The term, diabetes mellitus, is used to describe a syndrome characterized by chronic hyperglycemia and other disturbances of carbohydrate, fat and protein metabolism. Millions of people all around the world are suffering from diabetes mellitus with 5 – 10 % of them suffering from type I diabetes mellitus (otherwise known as insulin-dependent diabetes mellitus, IDDM) and 90 – 95 % of them from type II diabetes mellitus (also known as non-insulin dependent diabetes mellitus, NIDDM). While the most suitable and commonly available treatment for type I diabetes mellitus is insulin, there are number of oral drug therapies available for the treatment of type II diabetes, e.g., biguanides and sulphonylureas. The present study is essentially focused on glipizide which is a second generation sulfonylurea [3]. It contains not less than 98 % and not more than the equivalent of 102 % of I-cyclohexyl-3-{4-((2-(5-methylpyrazine-2carboxamido) ethyl)) benze-nesulphonyl} urea, calculated with reference to the dried substance [4]. Glipizide is widely used in the management of type II diabetes and, like other sulphonylureas, its overall mechanism of action is unknown. However, its predominant mechanism of action appears to be by increasing the secretion of insulin from the islets of Langerhans in both normal and diabetic humans and by decreasing the metabolic clearance rate of insulin [5]. Other proposed mechanisms include, increasing sensitivity of peripheral tissues to insulin effects, increasing the number of insulin receptors and increasing binding to and/or affinity of insulin for its receptors. Glipizide is almost completely absorbed after oral administration [6] but its absorption is delayed when taken with food. Therefore, glipizide is most effective when given at least 30 min before meals both in normal and diseased state [7]. Its initial dose is 5 mg once or twice daily with a maximum daily divided dose of 40 mg [6,8]. Peak blood levels are usually reached within 2 to 4 h after oral administration with a half-life of 3 to 7 h and duration of action of about 10 h in healthy human subjects [9]. Glipizide has an apparent volume of distribution of 0.13 l/kg in diabetic patients [10] and is contraindicated for patients with severe liver and renal disorders [3]. The most commonly encountered side effects include nausea, vomiting, headache, weakness, skin rashes, aplastic anemia, visual disturbances, impaired hepatic function, hypoglycemia and coma [6,11] . The objective of this study was to obtain pharmacokinetic data for glipizide in healthy and diabetic human volunteers with a view to ascertaining if there was pharmacokinetic variability between these two groups of subjects. EXPERIMENTAL Experimental design This study was an open, single-dose, parallel design. Twelve elderly diabetic and twelve elderly healthy human male volunteers (see Table 1) were included in the study. All subjects signed a written informed consent form before commencement of the study. The Pharmacy Ethical Committee of Faculty of Pharmacy, The Islamia University of Bahawalpur approved the study protocol for the human experiments. Each volunteer received a single tablet dose of glipizide 5mg (Minidiab®, Pharmacia & Upjohn, Pakistan) orally at about 8:00am, thirty minutes before breakfast [9,10,12]. The tablets were administered with 200 ml of water and total water intake did not exceed 1500 ml during the 12 h study period. The subjects remained seated either on a bed or a chair for the next 4 h. Standard lunch and dinner, not exceeding 2000 kcal, were served 5 and 12 h, respectively, after taking the tablet [9]. During the study period, the volunteers were constantly monitored by a physician in the Bioequivalence Sampling Room, Faculty of Pharmacy and Alternative Medicine, The Islamia University of Bahawalpur, Pakistan. Selection criteria for volunteers Healthy volunteers: Human subjects with good health as determined by medical history, physical examination and laboratory data (hematology, serum chemistry, urinalysis, creatinine), and having age and weight ranging from 45 -55 years and 70 -90 kg, respectively, were included in the study. Persons, who on physical examination showed signs suggesting abnormality of any organ system, or suffering from any infection or had any clinically significant abnormal laboratory values, were also excluded from the study. Volunteers with a known history of hyperglycaemia and hypoglycaemia were similarly excluded from the study. Furthermore, volunteers who had donated blood or suffered blood loss of > 200 ml in the four weeks preceding the screening were not enrolled in the study. So also were persons having any history of alcohol or drug dependency or drug abuse, known allergy to sulfa drugs, congestive heart failure, pulmonary disease, renal insufficiency, use of tobacco products in last 2 years, or are using any medication which may act as inducer or inhibitor of hepatic enzymes [13]. Diabetic volunteers: The inclusion and exclusion criteria of diabetic volunteers were similar to those for healthy volunteers except that fasting blood glucose level must be > 130 mg/dl. Only those patients who maintained a fasting blood glucose level over 130 mg/dl on prescribed diet and those who were without complication of diabetes mellitus were included in the study. Age and weight ranges were the same as for healthy volunteers. Patients having a history of insulin therapy were also excluded from the study [13]. Sample collection A 20-gauge venous canula was inserted into the forearm of the volunteers for collection of blood samples. Blood samples were collected just before drug administration (zero time) and then at 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 3.5, 4.0, 5.0, 6.0, 8.0, 10 and 12 h after administration of glipizide tablet both for healthy and diabetic volunteers [9]. Blood (3 ml) was collected in heparinized tubes, centrifuged and the plasma separated was stored frozen at -20°C until analysed [14]. HPLC analysis of glipizide Mobile phase: The mobile phase consisted of acetonitrile : 0.01M potassium di-hydrogen phosphate buffer (pH 3.5) in a ratio of 35 : 65 [15] which was degassed by sonication. It was eluted at a flow rate of 1.5 ml/min and the effluent was monitored at a wavelength of 275 nm. Run time for each injection was 20 min. Standard solutions: An initial stock solution of glipizide was prepared by dissolving 100 mg of the drug in 100 ml of methanol: dichloromethane mixture (10 : 90). Standard solutions were obtained by diluting the initial stock solution with mobile phase [16] to give drug concentrations in the range of 0 -1500 ng/ml. The solutions were stable for 3 to 4 months when stored at 4°C, protected from light. Standard curve: A standard curve encompassing the anticipated range of plasma glipizide concentration was constructed. Blank plasma (500 µL) was spiked with glipizide drug solutions to give concentrations of 50, 100, 200, 400, 800, 1000, 1200, 1400 and 1600 ng/ml in a 10 ml glass centrifuge tube [16] followed by the addition of 1 ml of 0.05M HCl [14] and vortexed for 45 s. It was extracted with 3 ml of toluene [14] by vortexing for 45 s, and then centrifuged at 4000 rpm for 15 min. The tubes were allowed to stand for 10 min and the clear supernatant was transferred to another glass tube and concentrated to dryness with a sample concentrator using nitrogen. The dry residue was reconstituted by vortexing with 75 µL of mobile phase and 20 µl of it was injected into the chromatographic system [15]. Sample preparation Plasma samples (500 µL) obtained from healthy and diabetic volunteers containing glipizide were taken in 10 ml centrifuge tubes and 1 ml of 0.05M HCl was added. The samples were then treated as described above for spiked plasma used in preparing the standard curve. Data analysis Pharmacokinetic analysis was performed using the non-compartmental method of analysis with Microsoft Excel 2003. The parameters, Cmax (maximum plasma concentration) and tmax (time to reach maximum plasma concentration), were determined from the concentration versus time curve. The area under the plasma concentration -time curve (AUC) was calculated by the trapezoidal rule. Statistical analysis was carried out with Statistical Package for Social Sciences, version 15 (SPSS Inc, USA). Descriptive statistics was adopted to summarize the data and MannWhitney-U Test was applied as inferential statistics to observe association between various variables among the two groups. A p value less then 0.05 was considered as statistically significant. RESULTS The pharmacokinetic data are listed in Table 2. The value of Cmax was 465 ± 60 and 472 ± 56 ng/ml for healthy and diabetic volunteers, respectively, while that of Tmax was 2.00 ± 0.06 and 2.04 ± 0.74 h for healthy and diabetic subjects, respectively. AUC0-∞ for healthy and diabetic subjects was 1878 ± 195 and 1723 ± 138 ng h/ml, respectively. On the other hand, the AUMC0-∞ (area under the mean curve) was 10169 ± 1467 and 9510 ± 658 ng.h2/ml for healthy and diabetic volunteers, respectively. Mean resident time (MRT) for healthy volunteers was 5.24 ± 0.31 and for diabetic volunteers 5.63 ± 2.86 h. The value of Ke (elimination rate constant) for healthy and diabetic volunteers was 0.25 ± 0.02 and 0.24 ± 0.01 h-1, respectively, while t1/2 (half life) for healthy and diabetic subjects was 3.04 ± 0.27 and 2.98 ± 0.16 h, respectively. Volume of distribution (Vd ) for healthy volunteers was 0.15 ± 0.01 L and 0.17 ± 0.02 L/kg for diabetic volunteers while clearance (Cl) for healthy and diabetic volunteers was 0.59 ± 0.06 and 0.64 ± 0.05 ml/min/kg, respectively. For all the parameters, no statistical significance was found between healthy and diabetic volunteers at p < 0.05. DISCUSSION This study was aimed at determining whether there is any variability in glipizide pharmacokinetic in the diseased state. Few studies are available in the literature have addressed this aspect of glipizide pharmacokinetics. Based on the results obtained, it is evident that all pharmacokinetic parameters did not show statistical difference between normal and diseased (diabetics) subjects. The pharmacokinetics of a drug relates to its absorption, distribution, metabolism and excretion processes within the body. Variability in any of these processes will modify the target site concentration and its pharmacological effects. Physiological, pathological, genetic and/or pharmacological factors may contribute to these variations [17]. Any disease state that affects and changes the pathophysiologic condition of organs (especially liver and kidneys) may cause a significant variability in distribution, metabolism and excretion of drugs.Pathophysiological states of different organs are estimated by serum indicators, for example, serum albumin and creatinine levels. The baseline serum chemistry (serum albumin and creatinine levels) of diabetic volunteers did not demonstrate any major differences from that of healthy volunteers. The small but insignificant differences observed indicate that diabetes at this stage had not altered the functions of major organs (liver and kidneys). As a result no significant differences in pharmacokinetics of glipizide were observed in the present study. Although AUC and half life values showed a trend that appears to show that they were lower in diabetic volunteers, however, these pharmacokinetic differences are of less clinical importance. The plots in Figure 1 confirm the similarity of glipizide pharmacokinetics in healthy and human subjects. A close related study by Kradjan et al [13], which investigated the effect of age and disease on glipizide pharmacokinetics also reported insignificant differences with respect to these parameters. Several other studies [9,10,12,15,18-21] conducted on either healthy or diabetic human volunteers have buttressed the findings obtained in our study. CONCLUSION The study showed that at an orally administered dose of 5 mg, glipizide kinetics is not significantly different in healthy and diabetic subjects. Therefore, data obtained using healthy volunteers can be extended to diabetics. The observed minor variation in pharmacokinetics is of less clinical importance. Glipizide could be administered without adjusting the dosage and dose frequency in diabetics to attain desired therapeutic effects. ACKNOWLEDGEMENT The authors are thankful to Dr. Tariq Ansari for his help in recruiting the diabetic patients for the study. REFERENCES
Copyright © 2011 - Pharmacotherapy Group, Faculty of Pharmacy, University of Benin, Benin City, 300001 Nigeria. The following images related to this document are available:Photo images[pr11021t2.jpg] [pr11021f1.jpg] [pr11021t1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}