 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
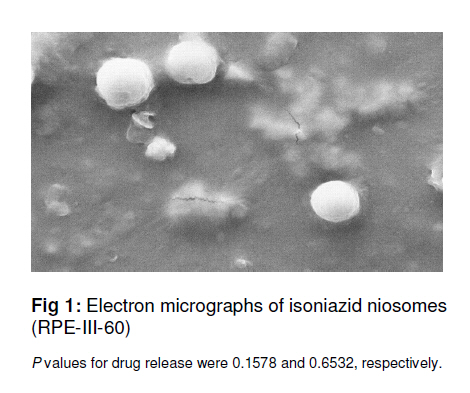
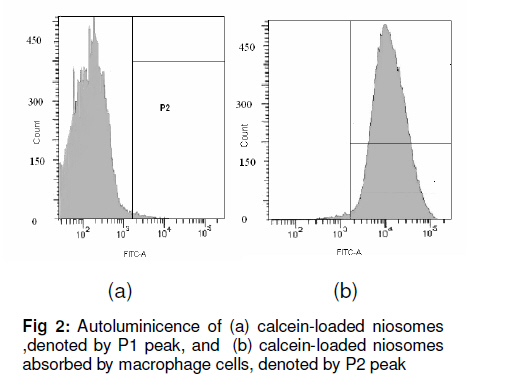
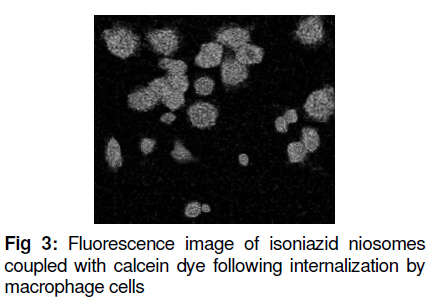
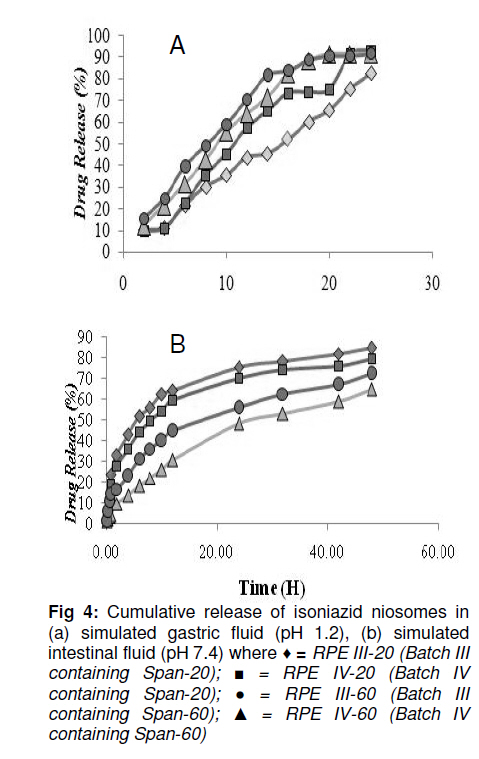
Tropical Journal of Pharmaceutical Research, Vol. 10, No. 2, April, 2011, pp. 203-210 Research Article Niosomal Delivery of Isoniazid -Development and Characterization Gyanendra Singh1, Harinath Dwivedi1, Shailendra K Saraf2 and Shubhini A Saraf1* 1Department of Pharmaceutics, Faculty of Pharmacy, Babu Banarasi Das National Institute of Technology & Management, Received: 10 September 2010 Revised accepted: 7 February 2011 Code Number: pr11028 Abstract Purpose: To develop a niosomal formulation for the delivery of isoniazid to achieve effective treatment of tuberculosis. Keywords: Niosome, Macrophage targeting, Isoniazid, Tuberculosis, Drug release, Cellular uptake. INTRODUCTION Isoniazid (INH) is an effective drug for the treatment of tuberculosis. There is evidence that isoniazid (INH) inhibits the synthesis of mycolic acid, an essential component of bacterial cell wall, and also combines with an enzyme that is uniquely found in strains of mycobacterium. Resistance to INH can occur due to reduction in intracellular penetration of the drug. Tuberculosis still remains the cause of high mortality and morbidity, particularly in developing countries. It is curable to a large extent but concern about the disease has resurfaced due to the emergence of multidrug resistant strains and the AIDS pandemic [1]. Short-term chemotherapy is associated with many disadvantages such as toxicity, decreased bioavailability at the target site and most important, non-compliance by the patient, thus leading to drug resistance. Some of the shortcomings of chemotherapy can be circumvented by developing a drug delivery system that would release the drug in a sustained manner [2]. The concept of using submicron carrier systems for the delivery of antibiotics has received recognition [3]. For effective chemotherapy, an optimal concentration of therapeutic agents must reach the affected tissue(s) but sometimes, in an attempt to achieve,this, serious side effects may occur. Niosome is a biodegradable, biocompatible and non-immunogenic carrier in which drug can be encapsulated. Encapsulation of bioactive agent in niosome prevents inactivation of the drug, targets the biologically active compound to the targeted tissue and provides slow release of the loaded drug into the circulatory system, thereby reducing its toxicity if effective uptake is achieved [4]. In earlier studies, antitubercular drugs such as INH and rifampicin (RIF) entrapped in PLG polymer when injected as a single dose, resulted in sustained release of the drug for 6 -7 weeks into various organs of mice. The pharmacokinetics of PLG-encapsulated antitubercular drugs, administered either individually or in combination in mice, has been reported [5,6]. EXPERIMENTAL Chemicals and drugs Span 60 and Span 20 (sorbitan monostearate, MW 430.6 and 346.5, respectively), cholesterol (MW 386.7), dicetyl phosphate (MW 546.9), chloroform, diethyl ether, and isniazid (INH, MW 137.1) were all obtained from Sigma, St. Louis, USA, while RPMI 1640 nutrient broth was procured from Sigma-Aldrich. All the other reagents used were of analytical grade. Preparation of niosomes Drug-containing niosomes were prepared by reverse phase evaporation (RPE) method, frequently employed for the entrapment of hydrophilic drugs [7], using Span 20 or 60 (SP) as a non-ionic surfactant, cholesterol (CH) as an enhancer of niosomal membrane rigidity and di-cetylphosphate (DCP) as a negative charge inducer [8]. Niosomes with varying ratios of these materials were prepared. They were dissolved (10mg/ml) in a chloroform: diethyl ether (1:1) blend (10 ml) in a 100 ml round bottom flask to form the organic phase. An aqueous phase containing isoniazid (10mg/ml) was added, with the ratio of the organic phase to aqueous phase ratio fixed at 5:1. The flask was covered with parafilm to prevent evaporation of the organic phase and then sonicated with an ultrasonic bath sonicator (SN2P, Toschon Industries Pvt Ltd, India) at 50 °C for 5 min. A stable white emulsion was formed from which the organic solvents were slowly evaporated at 50 °C using a rotary vacuum evaporator until a thin film was formed on the walls of the flask. The film was hydrated with 10 ml of phosphate buffer solution (PBS, pH 7.4) to produce an aqueous niosomal suspension containing 100 mg isoniazid/10ml. This suspension was initially kept at 50 °C in a thermostatted water bath for 1 h and at room temperature overnight to obtain the drug-niosomes. Separation of free (un-entrapped) drug Free (unentrapped) drug was removed from the niosomes by centrifugation of the dispersion [8] at 14,000 rpm at 4 °C for 60 min in a refrigerated centrifuge (3-18 K,Sartorius, AG Weender landstrasse, Gottingen, Germany). The supernatant was removed and the pellet (residue) was resuspended in PBS. This process was repeated twice to ensure that free drug was completely removed. Particle size determination The mean size of the particles was measured by an optical microscope (CH20i BIMF, Olympus India Pvt Ltd) fitted with a camera (Yoko CCD Camera, Taiwan) [7]. The particle size distribution of each formulation was evaluated by determining the size of 100 randomly selected niosomes with the aid of Medical Pro software (version 3.0). Evaluation of zeta potential and polydispersity index (PI) The surface charge of the niosomes was determined by measuring the electrophoretic mobility of the niosomal particles using a zetasizer (MAL 1021384, Malvern Instruments Ltd, UK). Polydispersity index was determined as a measure of homogeneity. Values were obtained from the printed report of Malvern zetasizer which includes the percent intensity in terms of size distribution of niosomes and their respective sizes [8]. Small values of PI indicate a homogeneous population while high values indicate heterogeneity. Determination of drug entrapment efficiency Entrapment of isoniazid (INH) in the niosomes was determined using a previously reported method [8,9]. This method includes separation of unentrapped drug using Sephadex-75 minicolumn and then evaluating the entrapment efficiency by disrupting the vesicles with 0.1 % Triton-X100. The resulting mixture was centrifuged at 3000 rpm for 5 min, with the supernatant was decanted off and suitably diluted with PBS pH 7.4. The drug was estimated spectrophotometrically (UV-1700, Shimadzu Corporation, Kyoto, Japan) at 261 nm against PBS containing Triton-X100 as blank. Entrapment efficiency (EE) was determined relative to the original drug concentration according to Eq 1. EE (%) = (ED/TD)100 …………………… (1) where ED is entrapped drug concentration and TD is theoretical drug concentration (10 mg/ml). Scanning electron microscopy The morphology of the niosome was studied by scanning electron microscopy (SEM, Leo430, Cambridge, U.K.). Niosome samples were prepared by air-drying on an aluminum stub. The stubs were then coated with gold to a thickness of 200 to 500 A0 under argon atmosphere using a gold sputter module in a high vacuum evaporator. The coated samples were randomly scanned by SEM. In vitro drug release evaluation The in vitro drug release profile of the isoniazid niosomal formulations was assessed by a previously reported method [10]. The niosomal formulation was first centrifuged to remove un-entrapped drug. Next, 1 ml of the niosomal sample was packed into a dialysis tube, which in turn was placed in a beaker containing 100 ml of PBS (pH 7.4). The solution containing the dialysis tube (MW cutoff 10,000 Da) was stirred on a magnetic stirrer at 50 rpm at 37 ± 1°C. Samples were withdrawn at various time intervals over a period of 48 h. Each withdrawal was followed by replenishment of the release medium with an equal volume of fresh medium to maintain sink conditions. The samples were analyzed spectrophotometrically for drug content at 261 nm (Shimadzu 1601, Japan). The tests were carried out in triplicate. Assessment of the physical stability of niosomes Samples of niosomes were sealed in 30 ml clear glass vials, stored at 4, 30 and 40oC, respectively, for 90 days and assessed periodically for changes in particle size and residual drug content as previously described. Establishment and maintenance of cell line A mouse macrophage cell line (J744 A.1) was grown as a monolayer using RPMI 1640 media, without glutamine, but supplemented with 10 % fetal bovine serum (for nurturing the cell line). The media were also supplemented with 100 µg/ml each of penicillin and streptomycin, to suppress the growth of microorganisms. The cell lines employed in this study were maintained in a humidified incubator at 37 ± 2oC with 95 and 5% air and CO2, respectively. Cells were subcultured twice weekly by simply resuspending the cells using 0.2 and 0.025% trypsin and EDTA, respectively, and then replacing half of the cell suspension with fresh media. Adherent cells were grown to 80 % confluence in tissue culture grade flasks and then subcultured by discarding the used medium and leaving the cells which adhered to the bottom of the flask. Viable cell count Viable and non-viable cells were distinguished using a haemocytometer. For this purpose, trypan blue was used. Live cells exclude trypan blue stain, leaving them with a normal appearance under the microscope. Dead cells, however take up the stain, making them appear blue. An equal volume of cells and stain (0.4 % w/v) were mixed and applied to the bright-line haemocytometer. The number of cells counted (ignoring blue cells) was multiplied by 2 x 104 to calculate number of viable cells/ml, thus taking into account the dilution factor upon addition of trypan blue. Cell uptake studies Calcein-loaded niosomes were prepared to study their uptake in J744 A.1 mouse macrophage cells. Plain calcein-loaded niosome were assayed. The calcein-loaded niosomes were prepared by a similar method to that used for the drug-loaded niosomes with calcein substituted for the drug. The concentration of calcein used was 0.5 mg/ml in PBS. The calcein-loaded niosomes were purified from unloaded calcein niosomes with Sephadex G-75 column. Calcein-loaded niosomes (1 ml) were diluted in 1 ml RPMI 1640 media and added to monolayers of J744 A.1 cell lines (2 x 105), grown in 96-well culture plate and incubated for 240 min at 37 ± 2ºC. The cell monolayers were thoroughly washed in PBS and lysed in 0.75 ml of lysis buffer (PBS containing 0.1% Triton X-100). These cells were exposed to serial concentrations of the indicated dye (20 -0.5 µg/ml of control formulation and 7.5 -0.5 µg/ml of coupled formulation in 100µl of culture medium, RPMI 1640 supplemented with 5% heat-inactivated fetal bovine serum) over a 3-day incubation period at 37°C, 5% CO2, and 95% relative humidity. Their fluorescence was measured in lysis buffer extract using florescence activated cell sorter (FACS, Becton Dickinson, Gurgaon, Hariyana, India) and all the calculations were carried out with Cellquest software (version 5.2, BD Biosciences, Canada). Fluorescent microscopy Cells were cultured in 6-well Petri dish with 1.8 mm cover slips for 24 h. Calcein-loaded niosomes were added to the cell culture media RPMI 1640 at a concentration of 0.1 mg/ml. After incubation for 3 h at 37ºC, the cells were washed six times with PBS. The cover slips were put on slides coated with buffered mounting medium and viewed with a Nikon fluorescence microscope (Japan) [14]. Statistical analysis: Data, including kinetic data, were processed with MS Excel 2007 and are presented as mean ± standard deviation. RESULTS Increase in noisome size was observed with increase in cholesterol content. In vitro drug release of RPE-III-60 was best explained by Higuchi equation as the plot showed the highest linearity (r2 = 0.98), followed by first order (r2 = 0.97) and zero order (r2 = 0.88). On the basis of regression coefficient (r2) and unpaired t-test, the highest r2 value was obtained for RPE-III-60. Hence, RPE-III-60 batch was selected for niosomal release experiments. Zeta potential for this batch was 23 mv and while polydispersity index was 0.14. These values indicate good stability and homogeneity of the formulation. The corresponding r2 (0.98) for the Koresmeyer-Peppas equation indicate good linearity while SEM revealed that the niosomes were spherical. Niosome formulations were more stable at 4 than at 30 and 40oC (75 ± 5% RH) (Figure 1, Figure 2, Figure 3). A value of 61.8 % was obtained as autoluminiscence of calcein-loaded niosomes absorbed by macrophage cells denoted by P2 peak. Figure 4 shows that as the proportion of cholesterol in the niosomes increased, particle size, drug entrapment and t50% (time taken to attain 50% drug release) also increased. An earlier study had also found that incorporation of cholesterol in niosomes delayed in vitro drug release [11]. The regression coefficient (r2) when the in vitro drug release data were subjected to Koresmeyer-Peppas, 1st order, Higuchi and zero order release models yielded 0.98, 0.98, 0.97 and 0.88, respectively for RPE-III-60. The release exponent, ‘n’, for the Koresmeyer-Peppas model was 0.78, which appears to indicate a coupling of diffusion and erosion mechanisms. This is the so-called anomalous diffusion mechanism, which indicates that drug release was controlled by more than one process. A value of n = 0.45 indicates Fickian diffusion; 0.45 < n < 0.89 indicates anomalous (non-Fickian) diffusion. Cell uptake of niosomes Cell uptake data indicate that calcein-loaded niosomes were taken up easily by J744A.1 macrophage cell line to a maximum level of 61.8 % for RPE-III-60. DISCUSSION Increase in particle size was observed upon increasing the cholesterol content. Entrapment efficiency increased to an optimum level on increasing the cholesterol level and thereafter decreased. Cholesterol (CH) is an important component for changing the fluidity of bilayers of niosomes as the molecules of the former are sandwiched between the molecules of the surfactant (SP) thus rendering the bilayer of the niosomes rigid [9]. On the basis of coefficient of regression (r2) data, formukation RPE-III-60 was selected as the optimum batch [12,13]. On the basis of r2 value, RPE-III-60 and RPE-IV-60 had correlation coefficient (R2) values close to one, i.e., 0.9454 and 0.8621, respectively. The corresponding plot (log cumulative percent drug release vs time) for the Koresmeyer-Peppas model indicate good linearity (r2 = 0.98). The release exponent ‘n’ was 0.78, which appears to indicate a coupling of diffusion and erosion mechanisms (or anomalous diffusion) and may indicate that drug release is controlled by more than one process[14]. At optimum SP:CH ratio (6:3), SP bilayers are saturated completely with CH molecules, hence no further significant increase in vesicle size was observed; however, drug entrapment was enhanced. Similarly, di-cetyl phosphate (DCP) also affected drug entrapment efficiency in the niosomes because its incorporation in the bilayers of the niosome enhances the vesicle size of the niosomes probably due to the identical charge in the bilayers, leading to repulsion in the bilayers and hence increase in size and drug entrapment. Negligible degradation and leaching was observed at temperature 4oC but significant leaching occurred at both 30 and 40oC, indicating the need to store the niosomes at 4oC. Zeta potential (ZP) of 23 mv and polydispersity index (PI) of 0.14 for formulation RPEIII-60 shows that the formulation was homogeneous since PI > 0.3 is indicative of heterogeneity [16]. Ideally, the optimum value of zeta potential should lie between +25 and 25 mv [17]. Outside this range, the niosomes may not remain stable for a long period of time and this could adversely affect parameters such as entrapment efficiency and sustained release. A higher zeta potential indicates higher kinetic energy and tends to move particles towards agglomeration [17,18]. Statistical analysis revealed that there was a significant difference between drug release in simulated gastric and intestinal fluids. On the basis of coefficient of regression (r2) data and unpaired t-test, RPE-III-60 batch with the highest Koresmeyer-Peppas r2 value was staken as the most suitable isoniazid niosomal formulation. The niosomes obtained in the present work released the entrapped drug at steady rate for a longer duration (20 -30 h) than the 10 – 15 h reported for gliclazide niosomes by other researchers [16]. Moreover, the niosomes prepared in our work were comparatively smaller and more homogenous than the gliclazide niosomes. The fluorescent studies indicate that the isoniazid niosomes are capable of remaining in treated sites in the body for prolonged periods while maintaining constant drug concentration in the process. However, studies to confirm the extent and duration of action of the formulation in vivo were not performed. Nonetheless, the isoniazid niosomal formulation should improve patient compliance as a result of reduced frequency of administration could lead to decreased dose-dependent side effects often associated with repeated administration of conventional isoniazid dosage forms. CONCLUSION Isoniazid niosomal formulations are capable of facilitating reduced drug dose, dose frequency and toxicity when given parenterally or via other suitable routes. Their use could improve patient compliance and most importantly, macrophage targeting would be possible at sites where the mycobacteria responsible for tuberculosis are harbored. ACKNOWLEDGEMENT One of the authors (Gyanendra Singh) wishes to acknowledge a scholarship grant from the All India Council for Technical Education (AICTE) during the course of this work. REFERENCES
Copyright © 2011 - Pharmacotherapy Group, Faculty of Pharmacy, University of Benin, Benin City, 300001 Nigeria. The following images related to this document are available:Photo images[pr11028f4.jpg] [pr11028f3.jpg] [pr11028f1.jpg] [pr11028f2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}