 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
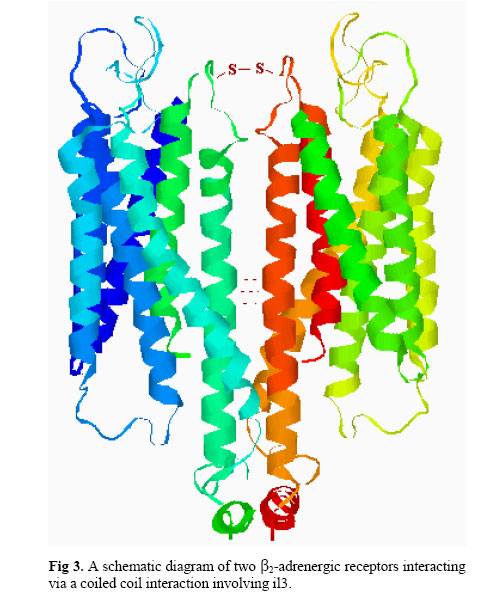
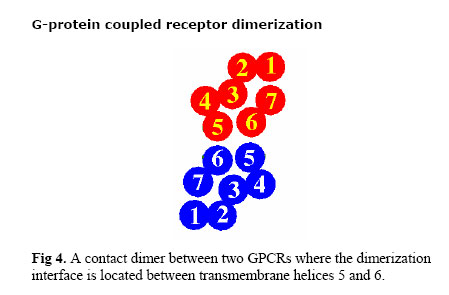
Iranian Journal of Pharmacology & Therapeutics, Vol. 2, No. 1, 2003, pp. 1-11 G-Protein Coupled Receptor Dimerization Nikzad Nikbin, Christine Edwards and Christopher A Reynolds Department of Biological Sciences, University of Essex,
Wivenhoe Park, Colchester, CO4 3SQ, United Kingdom Received January 28, 2003; Code Number: pt03001 ABSTRACT A growing body of evidence suggests that GPCRs exist and function as dimers or higher oligomers. The evidence for GPCR dimerization comes from biochemical, biophysical and functional studies. In addition, researchers have shown the occurrence of heterodimerization between different members of the GPCR family. Two receptors can interact with each other to make a dimer through their extracellular loops, transmembrane helices and intracellular loops. The nature of bonds between two receptors can vary from covalent (e.g. disulphide bonds) to non-covalent (for instance hydrophobic interactions between trans-membrane helices or coiled coil structures) or a combination of both. Dimerization can occur in and affect different stages of a receptor's life, namely trafficking, signaling and internalization, and can be seen as the natural way to regulate receptor activity or increase the functional repertoire of proteins. Different structures for GPCR dimers have been proposed, for example a simple contact dimer or an interlocking domain-swapped structure. Here we introduce some of the information available on GPCR dimerization, which includes early studies that had been dismissed until the relatively recent past and some of the more recent data which has vindicated these early studies. Keywords: GPCR dimerization, trafficking, signaling, internalization G-protein Coupled Receptors G-protein coupled receptors (GPCRs) are integral-membrane proteins, which face both the outside and the inside of the cell [1, 2]. The structural feature common to all GPCRs is the presence of seven hydrophobic transmembrane spanning a-helical segments that are connected to each other by intracellular and extracellular loops. The amino terminus is on the extracellular side while the carboxy terminus is intracellular (Fig 1). This family of membrane receptors comprises the largest superfamily of proteins in humans. More than 1000 different GPCRs have been identified in different organisms [3]. The endogenous ligands for this class of receptors are very diverse and include monoamines, peptides, glycoproteins, lipids, nucleotides and ions as well as exogenous stimulants such as light, odors and taste [3, 4]. On the basis of sequence homology, GPCRs are divided into different subfamilies: receptors related to rhodopsin (family A), those similar to the glucagon receptor (family B) and receptors related to the metabotropic neurotransmitter receptors (family C). There are also some minor subfamilies like those related to yeast pheromone receptors (family D or STE2 receptors and family E or STE3 receptors). Four different cAMP receptors in Dictyostelium discoideum comprise yet another subfamily (family F). As the name implies, GPCRs function through G-proteins. On the intracellular side a GPCR is connected to a heterotrimeric membrane-bound protein called the G-protein made up of three subunits: α ,β and γ . The α-subunit has the ability to bind guanyl nucleotides. When a ligand interacts with the receptor, a conformational change in the receptor is transferred to the G-protein. The α-subunit normally binds GDP in the resting state. Upon activation, GDP is released and replaced by GTP, which in turn imposes another conformational change on the G-protein and consequently the α-subunit dissociates from the βγ-subunits. Both the α-subunit and the βγ-dimer can now interact and activate a secondary messenger system such as adenylate cyclase. There are several types of G-proteins, such as Gs, Gi and Gq/11 which use different secondary messenger pathways. Once the receptor is activated, the intrinsic GTPase activity of the α-subunit converts GTP to GDP and the dissociated subunits recombine to form the inactivated receptor. GPCR signal transduction can also take place through other pathways that are not described here: the interested reader should refer to other articles [5]. GPCR Dimerization Until a few years ago, GPCRs were almost universally considered as monomeric and the relative stochiometry of receptor and G-protein was considered to be 1:1 [1, 5]. However, many experiments have challenged such a notion. Dimerization is known to be involved in the activation of single transmembrane receptors like growth factor receptors with intrinsic tyrosine kinase activity. These receptors, for example the receptor for epidermal growth factor, dimerize and autophosphorylate in response to interaction with ligands. Now, a growing body of evidence suggests that GPCRs exist and may even function as dimers or higher order oligomers. The evidence for GPCR dimerization comes from biochemical, biophysical and functional studies and is described below. In addition, possible roles for this phenomenon are discussed. Furthermore, the possibility of heterodimerization between different members of the GPCR family is described. Studies which show GPCR dimerization can be divided into two main categories: early pharmacological or biochemical evidence and more recent direct studies such as co-immunoprecipitation, biophysical methods, complementation experiments and even in one case x-ray crystallography. Pharmacological Studies Negative CooperativityOne of the earliest experiments which indirectly suggests that GPCRs might function as dimers is "negative cooperativity" between [3H]-(-)-alprenolol and alprenolol when interacting with the β-adrenergic receptor. As early as 1975, Limbird et al [6] suggested that a site-site interaction between two receptors (i.e. dimerization) could be responsible for the negative cooperativity observed in β-adrenergic ligands. To demonstrate the competition between two ligands, labeled [3H]-(-)-alprenolol was added to purified frog erythrocyte membrane containing β-adrenergic receptors. The receptors were in excess such that only a small minority of the sites were occupied. Dissociation of this drug-receptor complex was subsequently followed under two conditions: 1) at infinite dilution of the ligands such that no reassociation of the label will occur after dissociation and 2) in the same dilution in the presence of an excess of unlabelled (-)-alprenolol in such a way that the sites not occupied by [3H]-(-)-alprenolol will become saturated by the unlabelled drug. In the absence of co-operation between binding sites (which can be caused by dimerization), the rates of dissociation in both cases should be identical. However this was not the case. The increased rate of dissociation in the presence of excess unlabelled (-)-alprenolol indicates that filling of empty receptor sites acts on the sites already occupied by [3H]-(-)-alprenolol to accelerate dissociation. In other words the site-site interaction caused by receptor dimerization can lead to negative cooperativity. The same cooperativity was observed for m1 and m2 muscarinic receptors, suggesting that these receptors could function as dimers [7, 8]. In these studies, the Hill coefficient was less than 1.0, which could be interpreted as the existence of two binding sites for the same ligand; this gives another indication of dimerization. Radioligand Binding StudiesMore recent evidence for GPCR dimerization comes from radioligand binding studies. Ng et al [9] expressed human D2 dopamine receptors in insect Spodoptera frugiperda cells and immunoblotted them with a D2-selective antibody. A major band at 44 kDa was observed which could represent the D2 receptor monomer and another band at 90 kDa compatible with a dimer of the D2 receptor. Furthermore, they showed the benzamide D2 antagonist [3H]-nemonapride could bind to D2-dimers and monomers whereas the butyrophenone D2 antagonist [3H]-spiperone only binds to the monomer. In the next step, D2 dimers were incubated with peptides derived from the transmembrane domains (TM) of the D2 receptor, which resulted in dissociation of dimers to monomers. The same peptides were unable to dissociate dopamine D1 and serotonin 5-HT1B receptor dimers. The authors then concluded that receptor dimers are formed by specific intermolecular non-covalent interactions involving TM regions. Radioligand blotting studies also showed the presence of dimers for leutenizing hormone receptors [10]. Biochemical StudiesTarget Size AnalysisTarget size analysis and immunoaffinity studies suggested, as early as 1982, that the β 2-adrenergic receptor functional unit is two times heavier than its monomer [11]. The weight of mammalian β 2-adrenergic receptors was determined to be 59 kDa with monoclonal and autoantibody immunoaffinity chromatography with SDS-PAGE. The size of functional β2-receptor was then determined using target size analysis. According to radiation target theory, the biological activity of the protein is destroyed by a single high energy electron hit occurring within its molecular volume, so it is assumed that there is an inverse relationship between radiation inactivation of a protein and its size. Target size analysis of β2-adrenergic receptors suggested a functional molecular weight of 109± 5. The authors then concluded that the mammalian lung β2-receptor might be a dimer of two subunits of 59 kDa. In contrast to earlier studies (such as the negative cooperativity experiments) that used avian or amphibian erythrocytes, this study showed for the first time the possible existence of mammalian adrenergic receptor dimers. Using the same method, functional dimers were detected for a2-adrenergic [12], opioid [13], muscarinic [14] and gonadotropin releasing hormone receptors [15]. When the target size analysis was used to determine the size of the functional unit of the D1 dopamine receptor, the mass of the agonist-binding unit was higher than the antagonist one, which might imply that D1 agonists bind to receptor dimers (or induce them) and antagonists bind to the receptor monomer [16]. Cross-Linking StudiesCross-linking experiments were also used to show the existence of GPCR dimers. In this method, cross-linking agents such as gluaraldehyde, m-maleimidobenzoyl-N-hydroxysuccinimide ester (MBS) and dithiobis succinimidyl propionate are used to irreversibly capture receptor dimers. In such a study, cell membranes of PC-12 cells, which endogenously express bradykinin B2 receptors, were stimulated by bradykinin and receptor dimers were captured by the MBS cross-linker and detected using anti-bradykinin antibodies [17]. The authors proposed that the agonist bradykinin can induce dimerization, but the effect was not observed with the B2 antagonist HOE140. Using the same technique, the presence of receptor dimers was suggested for muscarinic receptors [18], calcium-sensing receptor [19], chemokine receptors [20] and dopamine D2 receptors [9]. In the same set of experiments [17], because addition of a peptide corresponding to the amino terminus of the receptor reduced the amount of detected receptor dimers, the involvement of the N-terminal of the B2 receptor in agonist-induced dimerization was proposed. In another set of experiments, cross-linking by radioiodinated agonists showed that the angiotensin II receptor could exist as a non-covalent dimer [21, 22]. Immunoprecipitation StudiesThe dopamine D3 receptor has been shown to exist as higher order oligomers using immunoprecipitation followed by Western blotting analysis [23]. In this study, membranes were prepared from human brain and incubated with a primary antibody (a D3-specific monoclonal antibody IgM). Bound antigen was detected using the appropriate peroxide-conjugated secondary antibody (for example goat anti-rabbit IgG) in conjugation with enhanced chemiluminescence [23]. This revealed the existence of three species of protein, one band showed a protein with molecular weight of 45 kDa (monomer) and there were two other species of 85 and 180 kDa (dimer and tetramer respectively). When the experiment was repeated with stable D3- expressing rat GH3 cells, only monomers were detected. An interesting observation made by the same researchers [23] was that when a naturally occurring truncated form of the dopamine D3 receptor, called D3nf is co-expressed with D3 receptor, using an anti-D3 antibody, D3nf can be detected in the immunoprecipitate suggesting dimerization between the D3 and D3nf proteins. Immunoprecipitation followed by Western blotting has revealed the possible dimerization for many GPCRs, including dopamine D2 [24], dopamine D1 [25], 5HT1b [26], the substance P receptor [27], the human C5a anaphylatoxin receptor [28] and the platelet activating factor receptor [29]. Although these results in cell lines were often interpreted as non-specific aggregation of incompletely folded intermediates, in each case the higher molecular weight species appeared to comprise multiples of the monomer, i.e. dimers or tetramers. However, the biochemical evidence for dimerization is not direct. To prove the specifity of dimer formation, research has focused on more accurate and direct methods. Immunoprecipitation Using Antibodies to Epitope-Tagged ReceptorsTo provide direct evidence for GPCR dimerization, Cvejic et al used differentially epitope-tagged opioid receptors in co-immunoprecipitation experiments [30]. A δ-opioid receptor tagged with the c-Myc epitope was co-expressed with the Flag epitope-tagged δ -opioid receptor. The expressed receptors were immunoprecipitated using anti-c-Myc antibody. Likewise, in Western blot analysis, anti-Flag antibody was used and receptor dimers were detected. When the same experiment was repeated for cells expressing only c-Myc tagged receptors, dimers could not be detected with anti-Flag antibody. This suggests that Myc-epitope-tagged and Flag-epitope-tagged receptors dimerize. So, immunoprecipitation with anti-Myc antisera followed by Western blotting with anti-Flag antisera can detect dimers. In the same study [30], the authors suggested that agonists can reduce the level of receptor dimers and this can lead to an increase in receptor internalization. This is different to what has been observed for other receptors. For example in the bradykinin B2 receptor [17], the presence of agonist increased the amount of detected dimers. The agonist could also increase the amount of detected dimers for β 2-adrenergic receptors [31]. Co-immunoprecipitation of differentially epitope tagged receptors was used to show the existence of dimers for other GPCRs including the histamine H2 [32] and calcium sensing receptors [33]. In another study, it was shown that co-immunoprecipitation could provide direct biochemical evidence to support the existence of β 2-adrenergic receptors [34]. When Myc- and HA- tagged β 2-adrenergic receptors were co-expressed, HA immunoactivity in fractions immunoprecipitated with anti-Myc antibody could be observed and this was taken as evidence of dimerization of β 2-adrenergic receptors. When HA-tagged-β 2-adrenergic receptor was co-expressed with Myc-tagged M2 muscarinic receptor, no dimer was detected, showing that the dimerization arose from specific b2-adrenegic interactions [34]. In co-immunoprecipitation studies, dimers are usually resistant to SDS denaturation and this might indicate the involvement of hydrophobic interactions in dimerization [35]. Detecting dimers in Living CellsSince GPCRs contain seven hydrophobic transmembrane domains, incomplete solubilization of receptors can lead to aggregation and this could be mistaken for dimerization, so one might consider the results of experiments such as immunoprecipitation as artifacts of solubilization. So, methods were needed to show the existence of GPCR dimers in living cells. To achieve this, biophysical assays based on light resonance energy transfer were used. Bioluminescence resonance energy transfer (BRET) is a naturally occurring phenomenon. BRET causes the fluorescence effect in several marine animals such as Renilla reniformis. BRET results from the nonradioactive energy transfer between luminescent donor and fluorescent acceptor proteins. For example in Renilla reniformis, the catalytic degradation of coelentrazine by luciferase (Renilla luciferase or Rluc) results in luminescence; this is in turn transferred to the green fluorescent protein (GFP), which emits fluorescence. There are two basic conditions for BRET to happen: first the donor and the acceptor should be in close proximity (like two protein subunits in a dimer) and second, the emission spectrum of the donor and the excitation spectrum of the acceptor must overlap. In one of the earliest GPCR experiments to use BRET, Angers et al [36], investigated β 2-adrenergic receptor dimerization. When fusions of β 2-adrenergic-Rluc and β 2-adrenergic-YFP (an enhanced red-shifted GFP) were co-expressed, addition of coelentrazine caused a broad bioluminescence signal. In addition to this, a fluorescence signal corresponding to the emission wavelength of YFP (530 nm) was observed, a sign of BRET occurring between two receptors as a result of receptor-receptor interaction. Because BRET can happen only if proteins are less than 50 Å apart, (for the parameters of the Angers et al study), dimerization (or a rearrangement of a pre-existing dimer) can be the only possible explanation for BRET. However it is important to note that BRET does not normally distinguish between dimers and higher order oligomers. To rule out the occurrence of BRET as an artifact of transmembrane protein overexpression, fusions of chemokine CCR5-YFP and β 2-adrenergic-Rluc were co-expressed; even when a higher level of receptor expression was applied, no BRET was observed [36]. The authors also ruled out the occurrence of BRET as a result of a spurious interaction between Rluc and YFP, because when they were expressed as non-fusion proteins, no BRET signal was detected. FRET (fluorescence resonance energy transfer) is another biophysical assay in which both donor and acceptor are fluorescent. Other methods include photo-bleaching FRET and time-resolved FRET. These methods have been used to show occurrence of dimerization in living cells for different GPCRs, including the d-opioid receptor [37], the thyrotropin-releasing hormone receptor [38] and the SSTR5- somatostatin receptor [39]. X-ray CrystallographyThe ultimate proof for the existence of GPCRs dimers may result from their solved three-dimensional structure. Due to technical problems this has not yet been achieved. However, the X-ray structure of the extracellular ligand-binding (amino terminal) region of the metabotropic glutamate receptor mGluR1 has directly shown the existence of dimers for this receptor [40]. Three different crystal structures of the extracellular ligand-binding (LB) region of this receptor were determined, one as a complex with the ligand glutamate and two structures as the unliganded free form [40]. All these three crystal structures showed a disulphide bridge between cys140 of two receptors in the dimer, however this bridge is located in a disordered segment of the protein. For the dimer interface, the author suggested helix-helix packing between α -helices B and C in each receptor (these helices are in the amino terminus not the transmembrane region). The authors suggested that movements of domains in the dimers might facilitate the separation of the transmembrane helices and intracellular regions and thereby activate the receptor. They also suggested the involvement of this "dimer activation"as a possible mechanism for activation of other GPCRs possessing extracellular ligand-binding sites [40]. Complementation (Functional Rescue) StudiesAlthough evidence for GPCR dimerization existed for many years,
some dating back to the early 70's, many researchers did not take it seriously.
One of the first studies that revitalized the idea of GPCR dimerization involved
co-expression of chimeric muscarinic/adrenergic receptors [41]
(see below under GPCR heterodimerization). In the same series of experiments,
a mutant muscarinic m3 (containing 16 amino acids of the m2 receptor
at the N terminus of the third cytoplasmic loop) when expressed alone was capable
of binding muscarinic ligands but it was functionally inactive. When this chimeric
receptor was co-expressed with another functionally impaired m3 receptor (P540A),
function was regained (Emax The same procedure was used to show dimerization of angiotensin II receptors [42]. When an angiotensin II receptor with a point mutation in helix 3 and one with mutation in helix 5 were co-expressed together, a normal binding site was restored despite the fact that none of the mutants when expressed alone could bind angiotensin II or different analogues [42]. No homologous recombination was detected and it was shown that the restoration of binding site was due to protein trans-complementation through receptor-receptor interactions. [42]. It is worth mentioning that even before the co-expression experiments, the split nature (domain structure) of some GPCRs had been shown [43] and the elegant work of Maggio et al [41] was based on this observation. In 1988 Kobilka et al [43] constructed ten chimeric receptors. These receptors were different combination of β2-adrenergic and α2-adrenergic receptors and it was shown that some of these chimeras were functionally active due to intermolecular interaction between receptors. Heterodimerization of GPCRsExperiments have shown the possibility of heterodimerization between different GPCR family members, for both closely or distantly related GPCRs. These experiments are reviewed here. Binding StudiesAs with the evidence for homodimerization, early evidence for receptor-receptor interaction between two different GPCR family members comes from binding studies. In 1980 Maggi et al [44] showed that when rat cerebral cortical slices were incubated with isoproterenol (a β-adrenergic agonist), an increase in α2-adrenergic receptor binding occurred. A decrease in binding of β-adrenergic ligands to their receptor was also observed. Most of the brain a-receptors are not located on nerve endings (presynaptic), so the authors suggested that modulation of a2-receptors using a β-adrenergic agonist could rise from intermolecular interactions between these two receptors located on the postsynaptic membranes [44]. A similar phenomenon has been observed for other receptors, such as adenosine A2/dopamine D2 [45]. The authors went on to consider this inverse reciprocal modulation as a possible mechanism for the homeostatic control of central noradrenergic activity. Complementation experimentsOne of the most convincing and direct pieces of evidence for GPCR heterodimerization comes from the elegant work of Maggio et al [41]. Two chimeric receptors α2/M3 and M3/α2 were created in which transmembrane domains 6 and 7 were swapped between the α2c-receptor and the M3 muscarinic receptor. These two chimeric receptors were expressed either alone or in combination and their ability to bind adrenergic and muscarinic ligands was investigated. Cells transfected with either of the two chimeras alone did not show any significant binding for muscarinic antagonist N-methyl scopolamine or adrenergic antagonist rauwolscine. When these two chimeras were co-transfected, binding sites for both ligands were detected. These binding sites had binding properties very similar to those of wild-type receptor except for a 5-fold decrease in the affinity for acetylcholine. The maximum number of binding sites (Bmax= 30-35 fmol/mg protein) was also lower compared to wild-type receptors (1 pmol/mg protein). In terms of being functionally active, when α2/M3 or M3/α2 were expressed alone and stimulated with carbachol no significant increase in intracellular IP1 level was observed. However co-expression of these receptors followed by exposure to carbachol resulted in a significant increase in IP1 levels; 83?15% above basal compared to 191?29% for wild type receptors. After co-expression, the EC50 calculated for carbachol (4.3?2.9 mM) was only 3-fold higher than that of wild-type M3 receptors. The authors concluded that ?cross-talk? between two chimeric receptors created functional M3 muscarinic and α2 adrenergic receptors. Because of the significant number of receptor sites and the fact that they showed 5-fold lower affinity for acetylcholine than the wild type receptor, the authors ruled out the possibility of accidental formation of wild-type receptors due to homologous recombination events (at the DNA level) [41]. Direct evidence for GPCR HeterodimerizationThe γ-amino butyric acid (GABA) metabotropic receptor (GABAB-receptor) has been known for several years. However, when this receptor was finally cloned, pharmacology studies showed that although it binds agonist, it lacks the expected signaling [46]. Publication of the GABAB-receptor sequence (now called GABABR1) led to identification of another related receptor that showed characteristics of family C GPCRs and was named the GABABR2-receptor. This second receptor was also functionally inactive when expressed alone. There are many lines of evidence that suggest GABABR1 and GABABR2 receptors function through heterodimerization. The two receptors have significant overlap in distribution and mRNAs encoding them can be found in cerebral cortex, hippocampus, cerebellum and other parts of the CNS. In addition when co-expressed, a 10-fold increase in agonist potency is observed compared to GABABR1 alone and finally, co-immunoprecipitation studies provide the direct evidence for heterodimerization [47, 48]. Furthermore, studies revealed that GABABR1 and GABABR2 function through creation of a heterodimer using a coiled-coil interaction between their intracellular C-terminus (Fig 2). In contrast to the GABAB-receptor, fully functional opioid receptors have also been shown to form heterodimers [49]. It had already been shown that δ-opioid receptors could form homodimers [30]: co-immunoprecipitation techniques had been used to show that κ -receptors also could exist as homodimers. However the properties of κ-opioid receptor homodimers were different to those of δ-dimers. For example, treatment of κ-receptors with agonist does not induce monomerization in contrast to δ-opioid receptors. To investigate possible interactions between these two receptors, Myc-tagged κ -receptors were co-expressed with Flag-tagged δ-receptors. Then antibodies specific for Myc-tag were used for precipitation. In the next step, anti-Flag antibodies were added to the precipitated material and Flag-tagged δ-receptors were detected. This clearly shows the intermolecular interaction between Myc-tagged κ-receptors and Flag-tagged δ-receptors. To test the selectivity of heterodimerization of δ and κ receptors, the same experiment was repeated for Myc-tagged κ-receptors and Flag-tagged m-opioid receptors. Under similar co-precipitation conditions, Flag-tagged m-receptors could not be detected [49]. Furthermore, when membranes of the cells expressing either δ or κ receptors were mixed, no heterodimerization was observed, suggesting that dimerization is not an artifact of extraction. κ-δ Heterodimers are destabilized when a reducing agent is used, implying that heterodimerization may result from disulphide bond formation between the two receptors. Other studies have also shown the ability of δ-opioid receptors to heterodimerize with μ receptors [50]. In addition to opioid receptors, heterodimerization has been reported to occur between other closely related members of family A GPCRs. For example using chimeric receptors, Scarselli et al [51] showed that D2 and D3 dopamine receptors can interact with each other to form a functional heterodimer which shows different pharmacological properties to those of wild-type D2 or D3 receptors. For example when D2 and D3 were "broken" in intracellular loop 3 (il3), four units were made: D2trunk and D3trunk (made of helices 1-5 and the N-terminal part of il3) and D2tail and D3tail (the C-terminal part of il3 and helices 6 and 7). It was shown that in most cases D3trunk/D2tail had a higher affinity for most agonist and antagonists compare to the wild type D2 and D3 dopamine receptors. Another difference is that these heterodimers are able to inhibit adenylate cyclase types 5 and 6 whereas the D3 receptor is not able to do so. In the case of the dopamine D2 receptor, although it inhibited adenylate cyclase 6, the IC50 was different to that of the D2/D3 heterodimer (2.05±0.15 nM compared to 0.083±0.011 nM for the heterodimer [51]. It has also been suggested that heterodimerization can happen between different subtypes of serotonin receptors [52]. What was interesting in these studies was that the integrity of the third cytoplasmic loop is not important, at least when the receptor is membrane-bound, for maintaining the correct fold and function of the receptor. This has been seen in many GPCRs including dopamine, α 2-adrenergic, M2 and M3 muscarinic, rhodopsin, vasopressin V2, GnRH and neurokinin NK1 [51], suggesting that the split nature of receptors and complementation resulting from dimerization is a general feature in this family. Even distantly related members of GPCRs are capable of forming heterodimers. Using co-immunoprecipitation methods and cells co-transfected with dopamine D1 and adenosine A1 receptors, Gines et al [53] showed that these receptors can form functionally interacting heterodimers. When A1 adenosine and D1 dopamine receptors were co-expressed in rat fibroblast cells, the antibody against A1 was able to co-immunoprecipitate D1 receptors, indicating a possible protein-protein interaction. A1 adenosine receptors were not able to form heterodimers with the D2 dopamine receptor when the same procedure was repeated with these two receptors, suggesting specificity of the D1/A1 heterodimer [53]. Immunoprecipitation using antibodies against epitope tagged receptors showed heterodimerization occurs between opioid receptors and β 2-adrenergic receptors [54]. Other distantly related GPCRs that appear to make heterodimers include D2 dopamine and somatostatin receptors [55] and bradykinin B2 and angiotensin II receptors [56]. Structure of GPCR DimersFor two members of the GPCR family to interact with each other, three sites could be involved: extracellular loops, transmembrane helices and intracellular loops. These regions can interact through covalent bonds (e.g. disulphide bonds) or non-covalent interactions (for example hydrophobic interactions between helices or coiled coil structures) (Fig 3). Each dimer structure is possibly stabilized through a combination of the above factors. For example, the only direct evidence for GPCR dimerization comes from the crystal structure for the metabotropic glutamate receptor mGluR1 N-terminal [40]. The data shows a disulphide bond between cys140 of each receptor N-terminal, however, because the location of these cysteine residues is ill defined, it was suggested that the dimer interface consists mainly of helical packing between α -helices B and C in each monomer. The existence of a disulphide bond in the mGluR1 dimer and its possible role in dimer stabilization was also concluded from co-immunoprecipitation studies [57]. Similar findings suggest the existence of disulphide bonding between extracellular loops 2 and 3 of the M3 muscarinic receptor [58] where site-directed mutagenesis studies showed that two conserved cys residues (cys 140 and cys 220) play a pivotal role in M3 dimer formation. In addition, it was shown that non-covalent bonds could also play a role in formation of M3 muscarinic dimers. A very interesting experiment conducted by Hebert et al [31] showed the importance of helix-helix interactions in dimer formation. It is a well known that some receptors consisting of a single transmembrane domain are activated through dimerization induced by agonist binding. Of these dimers, some are SDS-resistant including glycophorin A (GPA) in which the importance of the LIxxGVxxGVxxT motif has been shown for dimerization [59]. In this form of dimer, the transmembrane helices are believed to form a right-handed coiled-coil where non-covalent hydrophobic helix packing interactions dominate. The presence of Gly83 seems to be critical for GPA dimerization. Analysis of β 2-adrenergic receptor helix sequences revealed that such a motif exists at the cytoplasmic end of the sixth transmembrane helix [31]: 272LKTLGIIMGTFTL, where the placement of leucines and glycines is preserved in either direction. All these four residues are located on the external face of helix six and are available for receptor-receptor interaction. When this motif (residues 276-279) was synthesized, it was shown that addition of this peptide reduces the amount of β 2-adrenergic dimers by 69% after 30 min. A control peptide motif from helix seven of D2-dopamine had no effect on the amount of detected β2 dimers so the authors suggested that the dimer interface in the β2-dimer includes hydrophobic interactions from one receptor with helix six of another receptor and vice versa and this peptide could block the dimerization interface and so reduced the amount of dimers. It should be mentioned, however, that the GpA motif is not readily apparent in other GPCRs. Another possible site for intramolecular interaction is the intracellular loops. For example, coiled coil domains in carboxyl tails of GABABR1 and GABABR2 are suggested to play a role in GABAB receptor dimerization [60]. It should be mentioned that although a conserved coiled coil pattern does exist in the C-terminal of GABABR1 and R2, later reports [61] argued that this motif is important to mask the ER retention signal and if deleted, does not affect dimerization. The general requirement for a peptide to make coiled coil interaction with another peptide of the same or different sequence is the presence of a repeating seven-residue unit abcdef [62]. Residues a and d are large and hydrophobic and form a pattern of knobs and holes that interlock with those of the other peptide to form a hydrophobic core. On the other hand residues b, c and f which are located on the periphery of the coiled coil are usually charged and can face the solvent. Such a coiled coil motif also exists in the third intracellular loop of the b2-receptor as RFHVQNLSQVEQD. As can be seen, this peptide shows a pattern for a coiled-coil motif in either direction, starting from Leu as the first hydrophobic residue a. In both directions, residues d are valines, which fulfill the need for a largish non-polar residue in this position. Residues b, c and f are N, Q, R in one and S, Q, D in other direction which are polar. Such a conserved pattern might be involved in formation of a coiled coil structure between third extracellular loops of two adjacent receptors in a dimer (Nikbin, N., Reynolds, C.A. unpublished data). Possible Roles for GPCR DimerizationThe first fundamental question regarding GPCR dimerization is: why should dimerization be necessary for GPCRs? What is the evolutionary advantage of dimerization of an already complicated protein? Having seven transmembrane helices can provide enough "biological tools" for a protein to transmit a signal across a membrane. Dimerization can occur in three different stages of a receptor life: trafficking, signaling and internalization. There is evidence to support the involvement of GPCR dimerization in each of these three stages. Dimerization in GPCR TraffickingThe classical example of dimerization occurring during receptor trafficking comes from studies on GABABR1 and GABABR2 receptors [46-48]. It was reported that when GABABR1 is expressed, it remains intracellularly as an immature glycoprotein [63]. On the other hand GABABR2 can fold properly and transfer to the cell surface, but it cannot bind GABA or function properly. When these two receptors are co-expressed, they can both reach the cell surface as mature proteins and are fully functional. To explain this observation it has been suggested that GABABR2 acts as a chaperone for GABABR1, which has an endoplasmic reticulum retention signal in the coiled-coil motif in its carboxy tail. GABABR2 can mask this signal by forming a heterodimer with GABABR1 through a coiled-coil interaction of their carboxy tails [61]. In the case of V2 vasopressin receptors, it was shown that some mutants of this receptor could inhibit proper trafficking of the wild type receptor to the cell surface [64], through heterodimer formation. This shows transport of a receptor to the cell surface can strongly be affected by dimerization. Dimerization in GPCR FoldingThe role of dimerization in GPCR folding can be explained by investigating physicochemical properties of these proteins. Some members of the GPCR family have up to 1000 residues. This makes it inevitable for some polar residues to be exposed to the membrane environment, which is highly hydrophobic; this in turn destabilizes the system. Dimerization can be the answer to this problem. For example, it has been shown that a single Asn in a transmembrane helix in synthetic peptides provides a strong driving force for dimer formation [65]. Gln is shown to have an even higher potential than Asn to induce oligomerization [65]. From a protein structure point of view, function and folding dictates the occasional need for polar side chains. So although polar residues are usually buried in internal hydrophilic pockets of the protein, sometimes it is necessary to have a polar residue pointing toward the lipid bilayer to let the protein fold and function properly. This requirement increases as the protein becomes larger. The comparison of frequencies of occurrence of Asn and Gln in the population of single span membrane protein (0.2%) versus the multispan membrane proteins (1.3%) supports this assumption. [65]. In other words, for GPCRs to fold and function properly, more polar residues might need to be exposed to the lipid layer and one way to stabilize this destabilizing phenomenon is dimerization. When polar residues occur on the external face of GPCR helices, they are usually found in the parts near to extracellular or intracellular ends, close to the hydrophilic parts of the cell. Dimerization in Increasing Receptor FunctionalityThe other role for dimerization could be in expanding receptor diversity i.e. heterodimerization of different GPCRs can create a dimer with different pharmacological properties than either of the two monomers. For instance, the pharmacological properties of the κ-δ opioid heterodimer were different from those of each receptor expressed individually [49]. For example, κ-receptors have high affinities for their selective ligands e.g. agonist U69593 and antagonist norbinaltorphimine. Similar analogy applies to the δ-opioid receptors as they have high affinities for their selective ligands, agonist ([D-Pen2, D-Pen5]enkephalin; DPDPE) and antagonist (TIPPy). It was observed that a κ-δ heterodimer had no significant affinity for either of k or d selective ligands. On the other hand, this heterodimer had high affinity for partially selective ligands like the antagonist naloxone. Interestingly, the presence of a δ selective agonist (DPDPE) makes a κ-agonist (U69593) bind the heterodimer with high affinity. This synergy is also observed in the case of selective antagonists [49]. This clearly shows how receptor-receptor interactions can create new functional units with different properties to those of their components, hence expanding receptor diversity [49]. This idea is supported by the fact that the human genome contains fewer genes than what was thought about three years ago, so dimerization can be a way to increase the functional repertoire of proteins. Dimerization in ActivationDimerization can affect receptor activation and regulation. For example, dimerization might be the way that that body reduces the receptor response to an acute increase in endogenous ligands. For the adrenergic receptors, there is evidence to suggest that dimerization is induced when the concentration of agonists increases [6]. This in turn, dramatically decreases the affinity of the second site for the ligand. Another possible reason for the necessity of dimerization in activating GPCRs comes from the suggestion that the cytoplasmic surface of a rhodopsin monomer is only about half the size of transducin, the G-protein that couples to rhodopsin [66]. Accordingly, different receptors in a dimer interact with different domains or subunits of a G-protein heterotrimer. For example, one receptor could attach to Gα and the other to Gβγ to provide an efficient means of tilting Gα away from Gβγ and hence permit GDP release [66]. The same effect could be achieved by one receptor binding to the ras-like domain of Gα and the other receptor binding to the helical domain of Gα [67]. Receptor signaling has also been affected by dimerization. As described by Hebert [31], a peptide derived from transmembrane helix 6 of the β 2-adrenergic receptor can block the dimerization of this receptor [31]. When β 2-adrenergic receptors are treated with agonist, the number of dimers increases. On the other hand, the peptide from transmembrane helix 6 can decrease adenylate cyclase activity. Together, these observations indicate that the dimerization induced by agonist treatment leads the receptors to activation [31]. The fact that dimeric antibody against the second extracellular loop of the β2 adrenergic receptor can act as an agonist, supports this hypothesis and has even led to suggestions that dimerization is a prerequisite for receptor activation [68]. The GABAB receptor provides another piece of evidence to support a role for dimerization in receptor activation and signal transduction. As was stated before, GABABR1 has a retention signal in its carboxy tail. If this signal is deleted through mutation, the GABABR1 is capable of being transferred to the cell surface but is still not functional. This clearly shows the necessity of GABABR1-GABABR2 heterodimer formation for signaling [61]. Dimerization in InternalizationHeterodimerization can also affect internalization of different receptors, as shown by Jordan et al for the opioid homodimers [49] and opioid/adrenergic heterodimers [54]. It is known that etorphin, an opioid agonist, can induce internalization of d but not κ opioid receptors, when these receptors are expressed individually. When δ receptors are co-expressed with k receptors, etorphin cannot cause significant internalization and the researchers designated the observed 20% internalization to the occurrence of δ-homodimers. In another set of experiments, internalization of δ-opioid/β 2-adrenergic was investigated [54]. The fascinating result showed that when δ-receptors and β 2-adrenergic receptors are expressed together, δ-receptors internalized in response to β 2-adrenergic selective agonists. Radioligand binding studies ruled out binding of any δ2-agonists to δ-receptors, suggesting that internalization is a direct result of heterodimer formation, especially when it was shown that δ-opioid antagonists could not block this phenomenon. The role of heterodimerization in trafficking was further shown when β2-receptors were co-expressed with non-internalizing k receptors. β2-Adrenergic receptors in this experiment failed to internalize in response to adrenergic agonist. So, it appears that dimerization can significantly affect trafficking of GPCRs in addition to their signaling properties. Computational Studies of Possible Mechanisms of DimerizationTwo possible mechanisms for GPCR dimerization have been proposed so far. The first one is just a simple contact dimer in which two receptors locate next to each other (Fig 4) [67]. On the basis of complementation studies, a domain-swapped model for GPCRs has also been suggested [67, 69]. Either of these models could be held together by disulphide bonds [40, 57, 58]. Gouldson et al, based on experimental data as well as computational methods suggested a domain-swapped inter-locking model for dimers in which transmembrane helices 6 and 7 were swapped between two adjacent receptors (Fig 5) [69]. The dimer interface is proposed to lie between helices 5 and 6. This is consistent with Hebert's studies [31] in which a peptide derived from helix 6 could block β 2-adrenergic dimerization. Other helices like 2 and 3 have also been proposed to be involved in the dimer interface [69]. Based on the evolutionary trace method, a bioinformatics data mining approach to determining protein function from a multiple sequence alignment, Dean et al also found patches of conserved in class amino acids on the external face helices 5 and 6, which the authors suggested could form the dimer interface [67]. The evolutionary trace method is based on the idea that the location of functionally important residues (e.g. those involved in dimerization) is conserved and that these sites have a significant lower mutation rate. Other structures, e.g. involving a covalent link between transmembrane helices 4 [70] still need to be investigated. Concluding RemarksA growing body of evidence from biochemical, biophysical and functional studies suggests that GPCRs function as dimers, heterodimers and higher oligomers through interactions involving their extracellular loops, transmembrane helices and intracellular loops. The nature of bonds between two receptors can vary from covalent (e.g. disulphide bonds) to non-covalent (for instance hydrophobic interactions between transmembrane helices or coiled coil structures) or a combination of both. Dimerization can play a role in receptor trafficking, signaling and internalization and may be a way to modulate the functional repertoire of proteins. While GPCR dimerization is now fully accepted, there remain sufficient uncertainties in the precise structural form and function of the dimers to ensure that the field is still open to more future research. Note Added in ProofThere have been a number of recent BRET and FRET studies that continue to support the existence of dimers but appear to give conflicting results as to whether the proportion of dimer is affected by challenge with agonists and antagonists [71, 72] or unaffected (i.e. that dimerization is constitutive) [73, 74]. References
Copyright © 2003 by Razi Institute for Drug Research (RIDR) The following images related to this document are available:Photo images[pt03001f4.jpg] [pt03001f2.jpg] [pt03001f1.jpg] [pt03001f3.jpg] [pt03001f5.jpg] |
| |||||||||
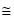 40-50 % of wild type m3,
Emax is the carbachol-induced phosphatidylinositol hydrolysis in
wild type receptors) upon stimulation with the muscarinic agonist carbachol.
This data was taken as direct evidence for intermolecular interaction between
muscarinic receptors. The authors also ruled out the accidental formation of
wild type m3 receptors due to homologous recombination (data not
shown here) [
40-50 % of wild type m3,
Emax is the carbachol-induced phosphatidylinositol hydrolysis in
wild type receptors) upon stimulation with the muscarinic agonist carbachol.
This data was taken as direct evidence for intermolecular interaction between
muscarinic receptors. The authors also ruled out the accidental formation of
wild type m3 receptors due to homologous recombination (data not
shown here) [{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}