 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
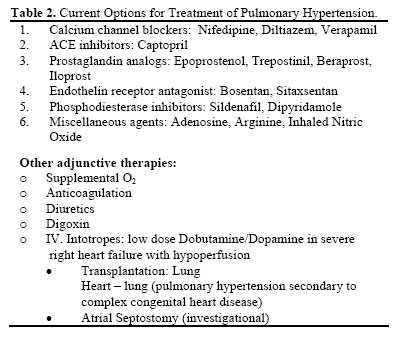
Iranian Journal of Pharmacology & Therapeutics, Vol. 4, No. 1, 2005, pp. 1-8 Recent Trends in the Management of Pulmonary HypertensionMANURU MUKHYAPRANA PRABHU, HASAN SREENIVAS MURTHY KIRAN, SUBISH PALAIAN and SUDHA VIDYASAGAR Department of Medicine (M.M.P.), Department of Pharmacology (S.P.), Manipal College of Medical Sciences, Manipal Teaching Hospital, Pokhara, Nepal; Department of Medicine, K.M.C. Manipal (H.S.M.K., S.V.), Manipal, India. Address correspondence to: Dr. M .Mukhyaprana Prabhu, Associate Professor, Manipal College of Medical Sciences, Manipal Teaching Hospital, Fulbari, Pokhara, Nepal. E-mail: drprabhu@fewanet.com.np Received December 16, 2004; Code Number: pt05001 ABSTRACT Context. Pulmonary hypertension is a common clinical problem encountered in day-to-day practice. Drug therapy forms the backbone of management and appropriate drug selection is based upon the underlying causes. The options available are widening with availability of newer therapeutic agents and thus it becomes necessary to be updated with the recent developments. Keywords: Primary and Secondary Pulmonary Hypertension, Treatment options, Prostacyclin analogues, Bosentan, Sildanafil Primary pulmonary hypertension (PPH) is a rare disease characterized by elevated pulmonary artery pressure with no apparent cause. Progressive obliteration of the pulmonary vascular bed is the hallmark of pulmonary hypertension (PH). The most common causes of pulmonary hypertension are related to pulmonary and cardiac diseases. Impaired pulmonary venous drainage from left heart failure, left ventricular diastolic dysfunction, and mitral valve disease are encountered most often. Another major cause of pulmonary hypertension, which may be reversible with long-term oxygen therapy, is chronic hypoxemia associated with structural or functional lung disorders [ 1] . Primary pulmonary hypertension refers to pulmonary artery hypertension with out an identifiable risk factor. Primary pulmonary hypertension is usually rapidly progressive, leading to right ventricular failure and death within a few years. The condition is nearly universally fatal, with few reports of disease regression. In patients with New York Heart Association (NYHA) class I disease (heart disease but no limitations on activity) or class II disease (symptoms with everyday activity), survival time is about 6 years. In those with class III disease (symptoms with less than everyday activity), survival is about 2.5 years, and in those with class IV disease (symptoms with any activity and sometimes at rest), survival is about 6 months. Overall, the 5-year survival rate of untreated primary pulmonary hypertension is about 20% [ 2] , with a median survival time of about 2.8 years [3] . Pathogenesis. Recent insights into the pathogenesis have resulted in newer approaches to its treatment. Pulmonary hypertension involves vasospasm and later intimal fibrosis, thrombosis in situ, proliferation of smooth muscles and medial hypertrophy. Normally there is a balance between the endothelium derived relaxing factors Nitric oxide (NO), prostacyclin and endothelium derived constricting factors (Endothelin-1 etc). Any imbalance of these factors promotes endothelial smooth muscle proliferation, induces vascular remodeling and incites thrombosis. Pulmonary hypertension is thus, nowadays, considered as a vasoproliferative rather than a vasoconstrictive disorder. This has led to a major change in the approach to treatment, which has evolved, from vasodilators to antiproliferative agents [ 4] . METHODSWe searched the MICROMEDEX database; updated version of June 2004 and Iowa Drug Information System (IDIS) updated version of September 2003 search for all published trials in this area. The search also included the indigenous journals to identify all published randomized controlled clinical trials evaluating the efficacy, tolerability and superiority of selected group of drugs. Few studies were located from the MEDSCAPE. Search terms included pulmonary hypertension, newer drugs for pulmonary hypertension, secondary pulmonary hypertension, ACE inhibitors, CCBs, prostaglandins, NO, arginine, PDE inhibitors, adenosine, endothelin receptor antagonists in pulmonary hypertension and other relevant terminologies. Studies were selected if efficacy, tolerability and superiority were reported as major outcome measures. The preference was given for the studies, which compared the superiority of drug(s) with that of other category ones. Typically the trials enrolled patients given the drugs and were followed up for 3 months to 5 years to establish a baseline reduction in Pulmonary Artery Pressure (PAP) and Pulmonary venous Resistance (PVR). The patients were either randomly assigned to get either placebo or study drug and being followed up for 6 months to 5 years while the efficacy in terms of PAP and PVR and tolerability were monitored. A responder rate is reported by measuring PAP and PVR in response to short acting vasodilators (e.g. inhaled NO, IV Adenosine or IV PGI2). A positive response is 10 mmHg or more fall in Mean Pulmonary Artery pressure (MPAP) with either no change or an increase in cardiac output and/or decrease in PVR of approx. 25%. TREATMENT OPTIONS FOR PULMONARY HYPERTENSIONIn this article we will attempt to summarize the results of clinical trials on new treatments and provide an approach to treatment of pulmonary hypertension. The diagnostic classification of Pulmonary Hypertension is given in Table 1. Current drug options for pulmonary hypertension are given in Table 2. The parameters for assessment of improvement used in various trials have been mentioned in Table 3. Calcium channel blockers (CCBs) Calcium channel blockers are vasodilators of systemic and pulmonary circulations. They act through the calcium channels located at the target sites. Among the various CCBs, long acting vasoselective dihydropyridines and slow release, heart rate regulating CCBs are preferred. Before starting long term CCBs, “responders” should be identified by measuring PAP and PVR in response to short acting vasodilators (e.g. inhaled NO, IV Adenosine or IVPGI2) [ 6] . A positive response is 10 mmHg or more fall in Mean Pulmonary Artery Pressure (MPAP) with either no change or an increase in Cardiac output and or decrease in PVR of approx. 25%. Rich et al [7] have shown that high doses of CCBs resulted in a 5-year survival of more than 90% in “responders” i.e. in patients who showed an acute response to vasodilators. It should be noted that only 10-15% of patients come under this “responder” category. The patients who respond to CCBs need large doses (eg. nifedipine up to 300 mg/day, diltiazem up to 720 mg/day). Woodmansey et al [ 8] reported oral administration of amlodipine achieved 20% or greater reductions in pulmonary vascular resistance and mean pulmonary artery pressure in 3 of 6 patients during a dose-finding investigation in patients with primary pulmonary hypertension or pulmonary hypertension due to thromboembolic events. Rich S et al in their study noted that administration of sublingual nifedipine 10 to 20 milligrams significantly increased cardiac output, and decreased mean aortic pressure, total pulmonary and total systemic resistance. An associated decrease in right ventricular enddiastolic volume and end-systolic volume was observed, as well as an increase in right ventricular ejection fraction. However, pulmonary artery pressure was not affected by nifedipine therapy. High dose nifedipine (up to 240 mg/day) or diltiazem (up to 720 mg/day) has shown to produce a reduction of 48% in mean pulmonary artery pressure and 60% in pulmonary vascular resistance in 8 of 13 patients [9]. The adverse affects are due to their negative inotropic effect and fall in systemic blood pressure, which may be an important limiting factor in their use. Hence CCBs should not be used in patients with overt right ventricular failure and those with systemic hypotension. ACE Inhibitors ACE inhibitors act by inhibiting the conversion of angiotensin-I to angiotensin-II and are widely used in treatment of systemic hypertension. Among ACE inhibitors, captopril has been used studied in pulmonary hypertension. Captopril. Captopril absorbed in its active form and is a sulfhydryl-containing angiotensin converting enzyme inhibitor. In its active form captopril is a highaffinity, competitive inhibitor of angiotensin converting enzyme (ACE; also known as kininase II) in human tissues. Although initial reports of captopril use in primary pulmonary hypertension (PPH) were positive, little research has been done sine the early 1980s. Recent reviews of management of PPH do not recommend angiotensin converting enzyme inhibitors in general or captopril specifically as particularly useful agents [ 10-11] . However the current status of ACE inhibitors in this area is not well known. Prostaglandin Analogs These agents act through an increase in Cyclic AMP, which produces vasodilatation. Epoprostenol. Epoprostenol is a potent pulmonary vasodilator. Its other mechanisms of benefit are unclear. It may also result in a positive inotropic effect, has some amount of systemic vasodilatation and antiplatelet effect by which it may reverse vascular damage [12] . Prospective randomized trials by Barst et al [ 13] and McLaughlin et al [14] have demonstrated improved exercise tolerance and survival in patients with primary pulmonary hypertension treated with epoprostenol. The survival benefit was seen in patients with NYHA class 3 & 4 heart failure. The optimal dose is yet to be clearly defined. However, starting epoprostenol at a dose of 2-4 ng/kg/min IV is recommended followed by increments of 1-2 ng/kg/min until either clinical improvement is seen or further increment is not possible due to side effects. Epoprostenol has a very short half life (t1/2 ). Therefore, a permanent central venous access is needed for administration and hence a risk of potentially serious complications thereof. Common side effects are flushing, headache, diarrhea, nausea, jaw pain and leg pain. Sudden withdrawal may result in rebound rise in PAP leading to acute RV failure. Epoprostenol is unstable in aqueous solution and has a t½ of 1-2 minutes. Hence any interruption of the infusion may lead to sudden life threatening loss of its effect. Therefore, a more stablePGI2 analogue like Iloprost offers theoretical advantages. Iloprost. Iloprost is a stable prostacyclin analogue with a t½ of 20-30min. Given intravenously (IV); its effects are similar to I.V. Epoprostenol. Dosing of IV iloprost is lower than that of IV epoprostenol. The normal starting dose is 0.5–1 ng/kg/min and maintenance dose is 2-4 ng/kg/min. Iloprost can be given by inhalation also. By inhalational route, less of the drug reaches systemic circulation thus making it a “pseudoselective pulmonary vasodilator”. Launay et al found aerosolized iloprost to be effective in improving NYHA class, exercise capacity, and subjective quality of life in 4 of 5 patients with pulmonary hypertension related to crest syndrome. Clinical and hemodynamic improvements were maintained in 2 patients for 2 years. No adverse effects were observed. One patient died due to severe right heart failure [15] . The combination of inhaled iloprost and oral sildenafil has also been studied in primary pulmonary hypertension. The combination was more effective than either drug alone in reducing mean pulmonary arterial pressure (PAP) in patients with primary pulmonary hyper tension. Wilkens et al [ 16] conducted a short-term study of 5 patients with severe pulmonary hypertension and New York Heart Association class III or IV disease, aerosolized iloprost (dose 8.4 to 10.5 micrograms) reduced mean PAP by 16% over baseline (p = 0.01), while sildenafil alone (50 to 100 milligrams) lowered PAP by 13% (p < 0.005). However, the combination reduced mean pulmonary arterial pressure by 25% (p = 0.02). There were no significant changes in heart rate or systemic arterial pressure during any treatment. Adverse effects were minor and transient. Trepostinil. The propensity for serious central venous catheter related infections in case of IV epoprostenol led to the development of trepostinil, a stable prostacyclin analog for subcutaneous infusion. It is given as a continuous subcutaneous infusion by a mini pump that has been used for insulin. It has a t½ of 3-4 hours. Simonneau et al [ 17] study showed that continuous subcutaneous trepostinil improved indices of dyspnoea, signs and symptoms of pulmonary hypertension and hemodynamics in primary and secondary pulmonary hypertension. Dosage used was 1.25 ng/kg/min-22.5 ng/kg/min thro subcutaneous infusion using micro infusion pump with catheter placed in the subcutaneous tissue. The most common side effect was infusion site pain (85%). In a large 12-week study involving patients with primary pulmonary hypertension (n = 271) or secondary pulmonary hypertension (n = 199) comparing placebo with continuous subcutaneous treprostinil showed significant improvement in exercise capacity at 12 weeks. Hemodynamic parameters monitoring showed 3 mmHg fall in pulmonary pressure 4 units/square meter (m2) fall in pulmonary vascular resistance index in treprostinil group whereas pulmonary pressure remained same whereas pulmonary vascular resistance index increased in placebo group [ 18]. The main benefits of treprostinil in short-term studies have been improvement of hemodynamics (eg, decreases in pulmonary arterial pressure and pulmonary vascular resistance index, increases in cardiac index), clinical symptoms (eg, Borg-Dyspnea score, Dyspnea-Fatigue rating, syncope), and quality of life [19- 23 ]. An improvement in NYHA heart failure functional class was observed in some studies [23] . However in the largest long-term study, 6% of patients discontinued treatment due to clinical deterioration [24] . Beraprost. Beraprost is a prostacyclin analog for oral use. Oral beraprost appears to be beneficial in the treatment of PPH [ 25- 26] . A study of beraprost in 13 patients with severe PH (given treatment for 1 year) concluded that beraprost may result in long-lasting clinical and hemodynamic improvement [25] . A placebo controlled multicenter trial (ALPHABET) of 130 patients showed that oral beraprost decreased Mean PAP in many patients. This study included patients with primary PH, PH associated with collagen vascular disease, congenital systemic to pulmonary shunts, portal hypertension and HIV. A median dose of 80 µg was given 4 times/day. Treatment with beraprost resulted in increases in exercise workload and peak oxygen consumption in a study involving patients with primary pulmonary hypertension (n=14) or chronic thromboembolic pulmonary hypertension (n=16). Beraprost was initiated at 60 or 80 micrograms/day (µg/day). The dose was increased over 1 to 2 weeks to the highest tolerated dose (mean 138 µg/day; range 60 to 360 µg/day) and divided into 3 or 4 doses daily for 1 to 7 months. Exercise testing was performed prior to and 3 months after initiation of beraprost. NYHA functional class in the beraprost group improved from 2.7 to 2.2 (p < 0.05). Beraprost increased peak workload from 87 to 97 (p < 0.001) and increased peak oxygen consumption from 14.9 to 16.8 ml/kg/min (p < 0.001) in patients with pulmonary hypertension. In addition, beraprost significantly decreased the ventilatory response to carbon dioxide production during exercise (p < 0.001). Side effects occurred in 53% of patients within 1 week and included flushing (20%), headache (13%), flushing plus headache (13%), diarrhea (3%), and nausea (3%). Further dose increments caused similar side effects in the remaining patients [27] . Limited studies suggest the efficacy of oral beraprost in infants and children with pulmonary hypertension secondary to congenital heart defects. Significant reductions in pulmonary vascular resistance (24% to 34%) were observed with usual doses of 1 µg/kg; the pulmo-nary-to-systemic resistance ratio decreased by 30% to 40% [ 28- 29]. Currently, IV prostaglandins are the first line of treatment for patients in NYHA class III & IV. However, in the future, patients with less severe disease (NYHA II, III) may be initially treated with one of the novel prostaglandins. Endothelin Receptor Antagonists Endothelin–1 mediates vasoconstriction and smooth muscle cell proliferation through ETA receptor. It also induces vasodilatation through ETB receptor. Bosentan. Bosentan is an oral, dual Endothelin Receptor antagonist. It blocks both ETA and ETB receptors (and hence it is non selective). It is thus an antiproliferative agent and may be most useful in PPH. A double blind randomized placebo-controlled study showed that oral bosentan increases exercise capacity and improves hemodynamics in patients with PH [ 30] . The results of BREATHE-1 [31] (Bosentan Randomized trial of Endothelin receptor Antagonist Therapy for Pulmonary Hypertension) concluded that bosentan is beneficial in patients with PH and is well tolerated at a dose of 125 mg twice daily. Elevation of liver enzymes transiently was observed in 14% patients an accumulation of bile acids in hepatocytes due to competitive inhibition of bile salt-export pump was the probable explanation. Sitaxsentan. It is an oral, selective ETA receptor blocker. A 12-week open label trial by Barst et al showed that sitaxsentan is beneficial in PH [ 32] . It was given orally at a dose of 100-500 mg twice a day. It also resulted in elevated liver enzymes in some patients. Phosphodiesterase Inhibitors Phosphodiesterases (PDEs) are responsible for the tissue levels of the cyclic nucleotides CAMP and CGMP. PDE inhibition may potentiate the vasodilatory effects of prostacyclins and NO. There are at least 11 isozymes of PDE with different affinities and tissue selectivities in mammals. PDE-3 and -4 are involved in the breakdown of CAMP metabolism, and PDE-5 for CGMP, but cross talk between CAMP and CGMP metabolism occurs. Sildenafil. PH is associated with a defect in endothelial NO production and hence NO dependent pulmonary artery dilation. Therapeutic approaches have been devised to reverse this relative deficiency of NO. This has been tried using L-arginine, the substrate required for NO synthesis and administration of NO as mentioned earlier. An alternative approach for enhancing the endogenous NO in PH is to increase NO–dependent, CGMP mediated PA vasodilatation by inhibiting the breakdown of CGMP by PDE5 (Phosphodiesterase 5). Sildenafil increases CGMP level by inhibiting PDE5, an enzyme that hydrolyses CGMP. There are few data on long term sildenafil treatment barring a few case repots. Studies that have been done so far are in a small number of patients. Michelakis et al [ 33] have demonstrated that sildenafil is as effective and selective a pulmonary Vasodilator as inhaled NO. Kothari and Duggal [ 33] have demonstrated that oral sildenafil is effective in PH. They studied 14 patients with severe PH (9 patients with primary PH & 5 patients were operated congenital heart disease patients). Oral sildenafil was started at low dose and then increased. It was used at a dose of 87.5 mg/day in children < 30 kgs and at a dose of 150 mg/day in patients > 30 kgs. They observed remarkable improvement in 6 min walk test, NYHA class and Mean Pulmonary Artery Pressure (MPAP). Bharani et al [34] studied 10 patients with PH of varied etiology (PPH, left to right shunts, pulmonary thromboembolism and interstitial lung disease) They used Sildenafil at a dose of 25 mg Q8H in their randomized double blind placebo controlled cross over study, and showed that Sildenafil was useful in PH of varied etiology. Ghofrani et al [ 35] , in a randomized controlled open label trial showed that Sildenafil causes preferential pulmonary vasodilatation and improves gas exchange in patients with severe lung fibrosis and secondary pulmonary HT. Sastry et al [ 36] studied the efficacy of sildenafil in 29 patients with PPH. Sildenafil was started at a dose of 25mg tid & increased up to 100 mg thrice a day. There was a significant improvement in functional class; 6min walk test and MPAP. In an open-label study of 12 patients with severe, non-operable chronic thromboembolic pulmonary hypertension, exercise capacity and hemodynamic parameters improved following oral treatment with sildenafil [ 37]. Ghofrani et al [ 38] in an open-labeled preliminary study reported sildenafil in combination with iloprost produces greater pulmonary vasodilatation than either agent alone in patients with severe pulmonary hypertension. No serious adverse effects occurred during combination therapy. The most commonly reported side effects of Sildenafil are due to vasodilatation. It can cause flushing, nasal congestion, headache, dizziness, hypotension. It can cause relaxation of lower esophageal sphincter resulting in dyspepsia and reflux related symptoms and transient visual abnormalities such as blurred vision and increased light perception due to inhibition of PDE 6 in rods and cones in retina [ 39] . It is contraindicated in patients with ischemic heart disease who are on nitrates. However, the experience with Sildenafil is limited and all are preliminary studies, more studies with control patients are required to establish its efficacy and safety. Dipyridamole. Dipyridamole, a phosphodiesterase inhibitor acts via increase levels of cyclic AMP in platelets. These effects potentates the platelet de-aggregating effects of prostacyclin, which also increases cyclic AMP. The effect of dipyridamole may not be due only to its effect on phosphodiesterase, but also on its ability to increase prostacyclin production and by its ability to inhibit adenosine metabolism or to inhibit erythrocyte uptake of adenosine. Dipyramidal in doses 0.6 mg/kilogram is effective in reducing pulmonary hyper tension in children [40] and pulmonary hypertension of newborn [41] when used alone or used with inhaled nitric oxide. Miscellaneous Agents Nitric oxide (NO). Nitric oxide (NO) is a reactive gas used via inhalation to produce selective pulmonary vasodilatation and improve oxygenation in patients with various forms of pulmonary hypertension. It acts through cyclic GMP. Inhaled NO is an effective acute pulmonary vasodilator in PH. Though, NO is a potent and selective pulmonary vasodilator, its long-term use is limited by its short t1/2 and needs to be given in continuous inhaler device. Inhaled NO is also currently used to identify “responders” (who respond to CCBs). Eighty ppm of NO is usually the maximum dose used in acute vasodilator setting [ 42] . Long-term therapy with NO is not practicable/viable at present. Nitric oxide acted as a selective pulmonary vasodilator and improved oxygenation in patients who underwent cardiac surgery and at risk for pulmonary hypertension [43] from oxygen requirement. NO was effec tive in maintaining pulmonary circulation without difficulty or complications in post cardiac surgical patients [43] . NO inhalation therapy should be considered in patients who developed hypoxemia after coronary artery bypass surgery [ 44] . Inhaled nitric oxide 10 to 80 ppm has effectively lowered pulmonary vascular resistance in infants and children with congenital heart disease and pulmonary hypertension, improving systemic arterial oxygenation [ 45]. Carmona et al [ 46] study on 14 patients with mild pulmonary hypertension after cardiac surgery, minimum airway resistance increased significantly at 60 minutes of nitric oxide (NO) inhalation (20 ppm) and remained elevated after NO was discontinued. L-arginine. Nitric oxide is synthesized from the amino acid L-arginine. Hence L-arginine supplementation may be beneficial in PH. Oral arginine decreased mean pulmonary arterial pressure and pulmonary vascular resistance and improved exercise capacity of patients with pulmonary hypertension when compared with placebo [47] . Oral ariginine was effective whereas, Intravenous infusions of L-arginine hydrochloride 12.63 g did not improve pulmonary hemodynamics in 4 patients with primary pulmonary hypertension [ 48] . Moreover, aortic oxygen tension decreased by 10 to 15% in three patients and severe hypotension occurred in two patients Adenosine. In patients with pulmonary hypertension, adenosine in doses 50 µ/kg/min appears to be an effective selective pulmonary vasodilator following cardiac surgery. Adenosine produced a significant reduction in mean pulmonary arterial pressure (22%), Tran pulmonary gradient, and pulmonary vascular resistance without changing systemic arterial blood pressure. The pulmonary vasodilatation produced by adenosine was associated with a significant increase in cardiac output. Pulmonary vasodilatation was achieved without a change in heart rate, intrapulmonary shunt fraction, or systemic arterial pressure [ 49] . Warfarin. Pathologic studies suggest that in situ thrombosis is a major contributor to progression of primary pulmonary hypertension [ 50- 51]. Patients with PH are at an increased risk of thrombosis. Reports suggests the positive role of warfarin in prognosis of primary pulmonary hypertension [52] . Long term warfarin therapy is also known to improve the survival rate [53] . The recent ACCP guidelines recommend anticoagulation with warfarin for patients with IPAH. The target international normalized ratio (INR) in patients with Idiopathic Pulmonary Artery Hypertension (IPAH) treated with warfarin is approximately 1.5 to 2.5. Anticoagulation of patients with PAH occurring in association with other underlying processes, such as scleroderma or congenital heart disease, is controversial. When deciding whether or not to anticoagulate patients with PAH occurring in association with underlying processes, the risk/benefit ratio should be carefully considered. It is generally thought that the risk of GI bleeding may be higher in patients with PAH occurring in association with scleroderma. Patients with PAH occurring in association with congenital heart disease may be at some increased risk of hemoptysis. However, patients with significant right-to-left intracardiac shunting may be at increased for paradoxical embolism to the CNS. Patients with portopulmonary hypertension may be at increased risk for GI bleeding due to the presence of varices. Generally, patients with PAH receiving therapy with long-term IV epoprostenol are anticoagulated in the absence of contraindications, due in part to the additional risk of cathe- ter-associated thrombosis [ 54]. COMMENTSWhat are the recent developments in the treatment of secondary pulmonary hypertension? Present treatment includes long-term oxygen therapy for COPD, and treatment of underlying diseases. Oral bosentan and subcutaneous trepostinil are not restricted for the use in only primary pulmonary hypertension. They have been approved for other causes of PH as well [ 55]. Sildenafil also has been shown to be useful in patients with severe lung fibrosis and secondary pulmonary hypertension [ 35] . Oral anti-coagulation is the mainstay of treatment for chronic pulmonary thromboembolism. What are the other existing adjunctive therapies for primary pulmonary hypertension? Long-term anticoagulation with warfarin is advised in all patients since it has been shown to reduce mortality by decreasing the incidence of intravascular thrombosis in situ. Supplemental O2 is useful in hypoxic patients. Patients with right heart failure should be treated with diuretics and digitalis. IV Dobutamine/Dopamine may be used for short-term treatment of severe right heart failure. Pulmonary Endarterectomy may be done in cases of chronic thromboembolic PH. Atrial septostomy – as palliative procedure can be tried in patients with severe precapillary PH but it is risky. How to approach a patient with pulmonary hy pertension [ 56-57 ]? There are no long-term data comparing different drugs. Hence the choice of drug therapy depends on experience, affordability and availability of drugs. All patients should be treated with oral anticoagulants if there are no contraindications. In patients with mild to moderate PH, in NYHA I, II and who come under the category of “responder” (following a response to acute vasodilator trial), calcium channel blockers are the first line of treatment. “Non Responders” in NYHA class II and who are stable may be observed. If necessary, oral beraprost or bosentan can be given. For patients in NYHA class III, oral/inhaled/subcutaneous prostalgndins or ET antagonist could be given as first line of treatment. For patients with severe PH (NYHA-IV) the treatment of choice is IV prostacyclin.
What are the future options? A gene for familial PH, which codes for BMPR-2, a receptor in the TGF-B family has been discovered. This is also seen in some patients with sporadic PH. This discovery may lead to specific therapies directed at the origin of the disease. There were no placebo-controlled trials prior to 1999. There are few studies evaluating the long-term efficacy of the drugs mentioned so far and their effects on survival. However, these drugs appear promising. Also, in future, a combination of 2 drugs from different groups may be tried. Trials of this kind are currently under progress, and hold out promises of hope for patients with irreversible pulmonary hypertension. CONCLUSION Pulmonary hypertension is a prevalent, serious medical condition that is treated largely by general practitioners, physicians and pediatricians. The development of newer drugs has expanded treatment options and offered significant benefits to patients, particularly the oral ones. Randomized controlled clinical trial data support the efficacy as well as tolerability of majority of these drugs. Also, in future, a combination of 2 drugs from different groups may be tried. Trials of this kind are currently under progress, and hold out promises of hope for patients with irreversible pulmonary hypertension. REFERENCES
Copyright © 2005 by Razi Institute for Drug Research (RIDR) |

{kind=link}
{kind=link}
{kind=link}